Sposoby rozdzielania substancji
Krystalizacja
Ten sposób rozdzielania jest bardzo powszechnie używany w laboratorium. Metodę tą wykorzystuje się
w chemii analitycznej jako ilościową metodę wagową. Krystalizacja może służyć do oczyszczania (wtedy nosi nazwę krystalizacji prostej) oraz do rozdzielania (znanej jako krystalizacja frakcjonowana) stałych substancji, które wytrącają się z odpowiednich roztworów.
Metoda ta bazuje na dwóch zależnościach. Pierwszą stanowi różnica rozpuszczalności związku, który ma być oczyszczony, a zanieczyszczeń, znajdujących się w roztworze. Zależność rozpuszczalności danego związku od temperatury to drugi proces wykorzystywany w krystalizacji. Zazwyczaj metoda ta opiera się na rozpuszczeniu zanieczyszczonej substancji w odpowiednio dobranym rozpuszczalniku w podwyższonej temperaturze. Potem roztwór należy gwałtownie schłodzić.
Nagłe ochłodzenie prowadzi do powstania roztworu przesyconego, który wytrącony ze stanu równowagi sprzyja krystalizacji określonego związku. Niestety objętość rozpuszczalnika nie pozwala na uzyskanie roztworu przesyconego względem zanieczyszczeń. Związki takie pozostają w ługu macierzystym. Najczęściej, jednak, gorący roztwór sączy się przez bibułę, na której osadzają się zanieczyszczenia (charakteryzują się mniejszą rozpuszczalnością w określonym rozpuszczalniku niż substancja oczyszczana).
Dobór rozpuszczalnika jest bardzo ważną kwestią, zależnie od rodzaju rozpuszczalnika wyróżnia się krystalizację z wody oraz z rozpuszczalników organicznych.
Rozpuszczalnik, który zapewni najbardziej efektywny rozdział to taki, który spełnia jedno z poniższych założeń:
- Substancja oczyszczana musi być bardzo dobrze rozpuszczalna w danym rozpuszczalniku na gorąco oraz równocześnie praktycznie nierozpuszczalna w niskiej temperaturze.
- Zanieczyszczenia muszą bardzo dobrze rozpuszczać się w określonym rozpuszczalniku w niskiej temperaturze, albo praktycznie nierozpuszczalne w gorącym rozpuszczalniku.
- Temperatura wrzenia danego rozpuszczalnika nie może przekraczać temperatury wrzenia oczyszczanej substancji.
Zanieczyszczenia są usuwane z roztworu, a następnie odsączane, w dwojaki sposób, zależnie od rodzaju rozpuszczalnika:
- Adsorpcja na węglu aktywnym pozwala na usunięcie zanieczyszczeń substancji krystalizujących z wody albo
- z roztworów alkoholowych.
- Adsorpcja na tlenku glinu, którą używa się podczas usuwania zanieczyszczeń z roztworów rozpuszczalników niepolarnych, ok. węglowodorów.
W niektórych przypadkach proces krystalizacji nie zachodzi zaraz po ochłodzeniu roztworu. Wytworzenie zarodków krystalizacji można osiągnąć poprzez wstrząśnięcie probówką lub zlewką z roztworem przesyconym, wrzucenie paru kryształów oczyszczonej substancji albo potarcie ścianek probówki lub zlewki bagietką.
Stopień czystości substancji stałej określić można mierząc temperaturę topnienia. Określona temperatura topnienia świadczy o tym, że mamy do czynienia z czystą substancją. Szerokie granice tej temperatury charakterystyczne są dla związków zanieczyszczonych. Obniżenie temperatury topnienia też jest oznaką występowania zanieczyszczeń. Takie zjawisko nosi nazwę depresji temperatury i stosuje się do porównania próbki substancji wykrystalizowanej oraz wzorcowej. Jeśli temperatura nie obniża się to wtedy uważa się, że próbka jest dostatecznie oczyszczona.
Preparatyka krystalizacji narzuca warunki bezpieczeństwa, jakie należy zachować, by nie paść ofiarą, tragicznego w skutkach, wypadku. Wiele z rozpuszczalników stosowanych często do krystalizacji jest palna
(z wyłączeniem wody, chloropochodnych, ok. CCl4 czy CHCl3. Praca z takimi rozpuszczalnikami musi być prowadzona w bezpiecznej odległości od ognia, ze względu na lotność substancji oraz intensywne pienienie mieszaniny po dodaniu węgla aktywnego (ze względu na dużą zawartość zaadsorbowanego powietrza). Najczęściej zabiegi związane z palnymi i wybuchowymi rozpuszczalnikami prowadzi się w tzw. pokoju benzenowym. Wiele związków posiada także właściwości toksyczne, dlatego też należy wystrzegać się wdychania par oraz krystalizacje przeprowadzać pod sprawnym dygestorium.
Do sprawnego przeprowadzenia krystalizacji, konieczne jest zastosowanie odpowiednich naczyń laboratoryjnych, sprzętu, a także umiejętności sączenia pod zwiększonym ciśnieniem, mierzenia temperatury topnienia. Przy wykonywaniu czynności laboratoryjnych bardzo ważne jest zachowanie odpowiedniej kolejności.
Krystalizacja acetanilidu z wody
Wykonanie ćwiczenia
W kolbie stożkowej płaskodennej umieszcza się 3g acetanilidu. Dadaje wodę destylowaną, ogrzewanie prowadzi się w płaszczu grzejnym, z kamyczkiem wrzennym (zapobiega przegrzaniu się cieczy). W przypadku nie rozpuszczenia substancji dodaje się porcjami wodę destylowaną (oczywiście nie przeprowadza się tego procesu w płaszczu grzejnym). Gdy cały acetanilid rozpuści się wsypuje się do roztworu ok. 0,5g węgla aktywnego,
a następnie doprowadza do wrzenia i gotuje przez 3 min.
Mieszaninę sączy się na gorąco przez karbowany sączek do kolby płaskodennej. Sączek uprzednio ogrzewa się w suszarce. Następnie przykrywamy kolbę korkiem albo parafiną i zostawiamy do krystalizacji. Kiedy roztwór ochłodzi się dalszy proces przeprowadza się w lodzie. Zestaw do sączenia pokazany jest na Rys. 1.

Rys. 1. Zestaw do sączenia roztworów wodnych
Krystaliczny osad sączy się na lejku Buchnera albo Schotta. W chemii analitycznej, przy przenoszeniu ilościowym na lejek stosuje się ług macierzysty, czyli przesącz z kolby ssawkowej. Następnie przesączone kryształy przemywa się zimną wodą destylowaną (kolejne porcje wody należy wlewać przy odłączonym ssaniu). Potem suszymy kryształy na lejku przy użyciu pompki wodnej. Osuszony osad umieszcza się w zważonej uprzednio szalce Petriego. Dla oczyszczonego acetanilidu określa się temperaturę topnienia i porównuje
z wartościami tablicowymi.
Krystalizacja nitroaniliny z etanolu
Wykonanie ćwiczenia
W suchej okrągłodennej kolbie umieszcza się 3g nitroaniliny oraz wlewa nieznaczną ilość etanolu. Dodaje się, podobnie jak przy pierwszym ćwiczeniu, kamyczek wrzenny. Ten rodzaj krystalizacji, z rozpuszczalnika organicznego wymaga specjalnej aparatury, konieczne jest zastosowanie chłodnicy zwrotnej. Sposób montażu przedstawiono na Rys.2.

Rys. 2. Aparatura do krystalizacji z etanolu (i innych związków organicznych, jako rozpuszczalników)
W przypadku nie rozpuszczenia całości nitroaniliny dodaje się alkohol etylowy przez chłodnicę. Po całkowitym rozpuszczeniu wlewa się dodatkowo 10% całkowitej ilości etanolu. Do gorącego, ale nie wrzącego roztworu, wsypuje się 0,5g węgla aktywnego (przedtem należy zdjąć chłodnicę). Następnie mieszaninę ogrzewa się jeszcze przez 3 min.
Mieszaninę sączy się na gorąco przez karbowany sączek do kolby płaskodennej. Sączek uprzednio ogrzewa się w suszarce. Następnie przykrywamy kolbę korkiem albo parafiną i zostawiamy do krystalizacji. Kiedy roztwór ochłodzi się dalszy proces przeprowadza się w lodówce. Zestaw do sączenia pokazany jest na Rys. 1.
Krystaliczny osad sączy się na lejku Buchnera albo Schotta. W chemii analitycznej, przy przenoszeniu ilościowym na lejek stosuje się ług macierzysty, czyli przesącz z kolby ssawkowej. Następnie przesączone kryształy przemywa się zimną wodą destylowaną (kolejne porcje wody należy wlewać przy odłączonym ssaniu). Potem suszymy kryształy na lejku przy użyciu pompki wodnej. Osuszony osad umieszcza się w zważonej uprzednio szalce Petriego. Dla oczyszczonego acetanilidu określa się temperaturę topnienia i porównuje
z wartościami tablicowymi.
Opracowanie wyników
W wyniku krystalizacji acetanilidu z wody otrzymano 1,07g osadu. Dla tego procesu można wyznaczyć wydajność reakcji:
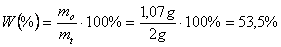
Określono temperaturę topnienia w przedziale 110 do 113 oC, podczas gdy wartość tablicowa jest równa 114 oC. Takie wyniki (depresja temperaturowa) świadczą o występowaniu zanieczyszczeń.
W przypadku p-nitroaniliny otrzymano 1,72g substancji krystalicznej. Zatem wydajność reakcji wynosi:
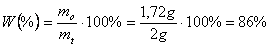
Zakres zmierzonej temperatury topnienia wynosił od 139 do 113 oC. Dość duży zakres oraz depresja temperaturowa są związane ze znacznym zanieczyszczeniem p-nitoaniliny.
Wnioski
Krystalizacja jest powszechnie stosowaną metodą oczyszczania związków organicznych. Niewątpliwymi jej zaletami są: prostota wykonania, małe nakłady finansowe, dużą wydajność procesu oraz wysoką skuteczność.
Uzyskane osady krystaliczne uzyskano ze stosunkowo małą wydajnością. Związki nie cechuje także czystość pozwalająca zastosować je jako odczynniki organiczne.
Destylacja
Metoda ta jest używana do oczyszczania (nosi wówczas nazwę destylacja prosta) albo rozdzielania substancji ciekłych (określana mianem destylacja frakcyjna lub refrakcja).
Destylacja opiera się na wykorzystaniu różnicy temperatur wrzenia cieczy rozdzielanych lub substancji ciekłej
i zanieczyszczeń (w przypadku stosowania tej metody do oczyszczania). Ciecz bardziej lotna ulega przemianie
w parę, a następnie jest skraplana. Związki nielotne pozostają w postaci ciekłej. W szczególnych przypadkach tworzą się roztwory, których nie da się rozdzielić przy pomocy destylacji, noszą one specjalna nazwę - roztworów azeotropowych.
Wydajność tej metody jest uzależniona od specyfiki aparatury, szybkości zachodzącego procesu oraz wielu innych czynników.
Temperatura wrzenia zależy ściśle od panującego ciśnienia zewnętrznego, jest właściwością fizyczną charakterystyczną dla określonej substancji. Pomiar dokładny jej wartości przeprowadza się w ebuliometrze. Pomiar temperatury wrzenia, poprzez umieszczenie termometru w cieczy, jest orientacyjny i różni się od wartości tablicowej. Kiedy chce się zebrać dana frakcję należy znać zakres temperatury wrzenia. Im mniejszy jest ten zakres, tym substancja ciekła jest czystsza. Stała temperatura wrzenia świadczy o braku zanieczyszczeń.
W praktyce zawsze mamy do czynienia z przedziałem temperatury wrzenia.
Pomiar zakresu temperatury wrzenia nie pozwala na określenie czystości frakcji. Jedną z najlepszych metod jest pomiar współczynnika refrakcji oraz porównanie jej z wartością tablicową. Jednak sposób ten nie może być stosowany we wszystkich przypadkach.
Bardzo ważne jest zwrócenie uwagi na bezpieczne przeprowadzenie destylacji. Wiele związków chemicznych jest palnych, duża ilość - toksycznych, a niektóre mogą spowodować wybuchy. Bezpośrednie ogrzewanie ciekłej substancji w kolbie może doprowadzić do przegrzania miejscowego (wskutek nierównomiernego ogrzewania), co powoduje pękniecie naczynia, a następnie do wywołania pożaru. Aby zapobiec takiemu zdarzeniu stosuje się chłodnice. Są one odpowiednio dobrane, powietrzne albo wodne, zależnie od wartości temperatury. Z takiego samego powodu używa się kamyczków wrzennych (stanowią je niewielkie kawałki fajansu albo porowatej porcelany).
Często stosowaną techniką jest także destylacja pod zmniejszonym ciśnieniem. Zmiana warunków destylacji pozwala na wykonanie rozdziału substancji ciekłych, które w normalnych warunkach są wybuchowe albo rozkładają się w temperaturze wrzenia bądź niższej.
Przeprowadzając destylacje należy uważać szczególnie na staranne zestawienie aparatury. Ważne jest także odpowiednie zebranie poszczególnych frakcji, a także prawidłowy pomiar współczynnika refrakcji (załamania światła).
Destylacja prosta
Wykonanie ćwiczenia
Do kolby okrągłodennej wlać 25 cm3 zanieczyszczonego etanolu, dodać kamyczek wrzenny. Potem należy zestawić sprzęt do destylacji prostej, którą przedstawia Rys. 3.

Rys.3. Sprzęt stosowany do destylacji prostej
Przed rozpoczęciem ogrzewania nie można zapomnieć o uruchomieniu chłodnicy wodnej. Ważne jest śledzenie zmian temperatury. Przy temperaturze niższej od temperatury wrzenia etanolu zbiera się tzw. przegon. Przy stabilizacji temperaturowej do osobnej elenmajerki zbiera się frakcję główną. Dodatkowo podczas wrzenia etanolu można zaobserwować znaczną akcelerację procesu. Alkohol etylowy zbiera się do momentu zmniejszenia temperatury, chociaż dla niektórych cieczy temperatura może się podwyższyć. Po otrzymaniu frakcji głównej znowu zbiera się pogon.
Po zakończeniu destylacji wyznacza się objętość alkoholu etylowego. Ponadto określa się zakres temperatury wrzenia, a także mierzy współczynnik refrakcji. Zgodność jego wartości z tablicową pozwala na określenie efektywności destylacji.
Destylacja frakcyjna (rektyfikacja)
Wykonanie ćwiczenia
Do kolby okrągłodennej należy nalać 20 cm3 mieszaniny heksanu oraz toluenu. Należy dodać kamyczek wrzenny oraz zestawić aparaturę zgodnie z Rys. 4.

Rys. 4. Zestaw do rektyfikacji
Do tego rodzaju destylacji również stosuje się chłodnicę wodną. Należy powoli ogrzewać ciecz, stale kontrolując temperaturę mieszaniny.
Zbiera się pięć frakcji. Określa się ich objętość, przedział temperaturowy, mierzy współczynniki refrakcji. Oblicza się także procentowy skład objętościowy poszczególnych frakcji, uwzględniając prostą wzorcową (wyznacza się ją na podstawie znanych współczynników refrakcji dla czystego heksanu oraz toluenu).
Opracowanie wyników
Zanieczyszczony alkohol etylowy został oddzielony dzięki metodzie destylacji prostej. Destylat zbierano
w zakresie temperatury 77 do 80oC. Porównując ten przedział, z wartością literaturową wynoszącą 78oC, można zauważyć, iż jest ona zbliżona do zmierzonej. Współczynnik załamania światła doświadczalny był równy 1,3587, natomiast wartość tablicowa wynosi 1,3594, tak więc obie liczny są porównywalne. Znając objętość alkoholu etylowego - 22,4 cm3 można wyznaczyć wydajność procesu destylacji:
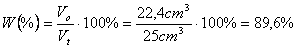
Przy pomocy destylacji frakcyjnej wyodrębniono toluen oraz heksan. Uzyskane wyniki: zakres temperaturowy zbierania destylatu, współczynnik załamania światła oraz objętość frakcji zebrano w Tabeli 1.
|
Lp.
|
Zakres temperaturowy
[oC]
|
Współczynnik refrakcji
|
Objętość destylatu
[cm3]
|
|
1
|
69 - 74
|
1,3785
|
4,8
|
|
2
|
74 - 80
|
1,4073
|
5,1
|
|
3
|
80 - 87
|
1,4528
|
4,7
|
|
4
|
87 - 96
|
1,4731
|
4,9
|
|
5
|
96 - 107
|
1,4913
|
5,0
|
Tabela 1. Wyniki destylacji mieszaniny toluen - heksan
Na podstawie znajomości współczynników refrakcji dla poszczególnych frakcji używając prostej wzorcowej można wyznaczyć procentową zawartość składników w kolejnych frakcjach:
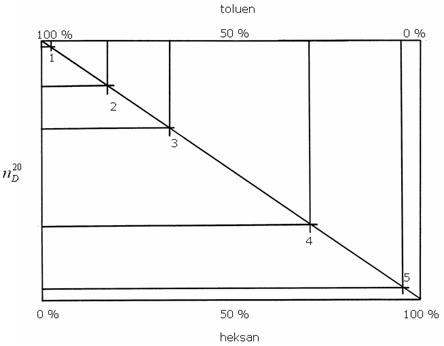
Prostą wzorcową opisuje zależność:
y = 0,0012x + 1,3726
Opierając się na w/w równaniu, uwzględniając wartości współczynników załamania światła wyznaczono procentową zawartość poszczególnych frakcji (Tabela 2):
|
Lp.
|
Zawartość toluenu
[%]
|
Zawartość heksanu
[%]
|
|
1
|
4,92
|
95,08
|
|
2
|
28,92
|
71,08
|
|
3
|
66,83
|
33,17
|
|
4
|
83,75
|
16,25
|
|
5
|
98,92
|
1,08
|
Tabela 2. Zawartość procentowa poszczególnych frakcji
Destylacja frakcyjna jest bardzo wygodną metodą, która służy do otrzymania różnego rodzaju mieszanin,
o innym składzie. W pierwszym oraz piątym przypadku otrzymane zostały prawie czyste składniki: odpowiednio heksan oraz toluen.
Wnioski
Destylacja czy to prowadząca do oczyszczenia cieczy, czy do rozdziału jest metodą bardzo skuteczną, o dużej wydajności. Pod warunkiem umiejętnego zestawienia aparatury charakteryzuje się prostotą oraz szybkością. Jej wadą, natomiast, są wysokie koszty, które uniemożliwiają, bardzo często, zastosowanie jej w produkcji.
Ekstrakcja
Ta technika rozdzielania mieszanin polega na przejściu substancji rozpuszczonej z jednego do drugiego rozpuszczalnika, przy czym musi być zachowany warunek niemieszalności wzajemnej obu rozpuszczalników. Ekstrakcja jest stosowana w przypadku, gdy do zmiany określonego rozpuszczalnika na taki, który będzie dogodniejszy lub, gdy zachodzi konieczność, selektywnego rozdziału mieszaniny. Do rozdziału bardzo często stosuje się rozpuszczalniki organiczne.
Różne rodzaje tej metody wymagają użycia innej aparatury. Proces rozdziału, który zazwyczaj używa się
w laboratorium, jest oparty na wytrząsaniu roztworu z czystym rozpuszczalnikiem w rozdzielaczu. Operacje tą powtarza się kilkakrotnie ze względu na osiągnięcie większej wartości współczynnika podziału pomiędzy obydwiema fazami. Potrząsanie zwiększa powierzchnie styku niemieszających się cieczy, a także, w znacznym stopniu, wzrost szybkości osiągnięcia termodynamicznej równowagi. Po każdorazowym wytrząśnięciu należy odczekać jakiś czas do ustalenia tej równowagi, a następnie rozdzielić fazy.
Bardzo ważnym zagadnieniem jest przestrzeganie podstawowych praw bezpieczeństwa. Wskutek różnego typu procesów w rozdzielaczu następuje zmiana ciśnienia (zazwyczaj wzrost), a to przy otwarciu korka doprowadzić może do pożaru, czasem wybuchu (jeśli rozpuszczalnik jest narażony na kontakt z otwartym ogniem), a także do oparzenia. Otwarcie kranika w rozdzielaczu umożliwia wyrównanie ciśnienia, a naczynie to powinno być trzymane w taki sposób, by wylot był skierowany w stronę ściany bądź tej części laboratorium, w której nie ma nikogo).
Ekstrakcja kwasu benzoesowego oraz benzoesanu etylu
Celem ćwiczenia jest rozdzielenie mieszaniny kwasu oraz jego soli. Ekstrakcja tych substancji następuje dzięki różnicy współczynników podziału dwóch faz: chlorku metylenu oraz roztworu wodnego wodorotlenku potasu. Ważną kwestią jest rozróżnienie obu faz, tzn. wodnej (nieorganicznej) i organicznej. W doświadczeniu wykorzystuje się relacje zobojętniania, dlatego należy zachować szczególną ostrożność. Oczyszczanie kwasu benzoesowego oraz benzoesanu etylu przeprowadza się przez krystalizację (kwas) i destylacje pod obniżonym ciśnieniem (sól).
Proces destylacji przy mniejszym ciśnieniu jest przeprowadzany podobnie jak w przypadku destylacji klasycznej. Niemniej jednak zwraca się większą uwagę na uszczelnienie sprzętu. Obniżenie ciśnienia można uzyskać przez zastosowanie odpowiedniej pompy, w tym przypadku dostaje się wartość ciśnienia ok. 15 mmHg stosując pompkę wodną. Taka metoda powoduje obniżenie temperatury wrzenia benzoesanu etylu. Należy również dodać, ze w tym przypadku nie używa się kamyczka wrzennego, ponieważ jego rolę przejmuje długa kapilara, której koniec dotyka dna kolbki.
Ze względu na pace w zmienionych warunkach ciśnieniowych należy:
- Stosować sprawdzone naczynia, wężyki, najlepiej takie, które mają odpowiednie certyfikaty
- Stosować kolby wyłącznie okrągłodennne
- Nosić okulary ochronne, przeznaczone do pracy ze żrącymi substancjami
- Przestrzegać zaleceń stosowanych przy używaniu pompki wodnej
- Zgłaszać osobie odpowiedzialnej za bezpieczeństwo nawet niewielkie usterki
Wykonanie ćwiczenia
Do roztworu, który należy rozdzielić dodać 50 cm3 chlorku metylenu, przelać całość do gruszkowego rozdzielacza (którego objętość nie jest mniejsza niż 250 cm3). Następnie dodaje się 15 cm3 uprzednio sporządzonego 5% wodnego roztworu wodorotlenku potasu. Po starannym wytrząśnięciu i ustaleniu stanu równowagi zleć warstwę dolną do kolby a, natomiast drugą do kolby b. Ekstrakcję roztworu z kolby
a przeprowadza się jeszcze dwa razy z użyciem wodorotlenku.
Wszystkie spuszczone roztwory b łączy się ze sobą, a następnie dodaje roztworu kwasu chlorowodorowego (1:1) aż do zakwaszenia takiego, by pH=1. Utworzony osad sączy się na lejku Buchnera albo Schotta, następnie przemywa wodą, suszy. W celu oczyszczenia przeprowadza się krystalizację, ponownie saczy na lejku Buchnera, suszy, przekłada na szalkę Petriego oraz wyznacza temperaturę topnienia.
Roztwór z kolby a przemywa się wodą destylowaną w rozdzielaczu, oddziela, następnie suszy nad bezwodnym siarczanem (VI) sodu. Oczyszczanie benzoesanu etylu przeprowadza się przy zastosowaniu metody destylacji przy obniżonym ciśnieniu. Należy zestawić sprzęt według Rys. 5., z uwzględnieniem nasmarowania części doszlifowanych smarem, starannego łączenia węży gumowych.

Rys. 5. Aparatura do destylacji pod obniżonym ciśnieniem
Następnie odsącza się środek osuszający, usuwa rozpuszczalnik - chlorek metylu poprzez odparowanie go na wyparce. Tak otrzymany roztwór przenosi się do kolby okrągłodennej i podłącza do zestawu destylacyjnego. Po włączeniu pompki wodnej, przy stałej kontroli kapilary, oczekuje się na ustalenie ciśnienia. Gdy osiągnie wartość 15 mmHg, ciecz należy powoli ogrzewać, kontrolując temperaturę. Podobnie jak przy destylacji pod ciśnieniem atmosferycznym, najpierw zbiera się porzegon, dopiero po ustaleniu wartości temperatury zbiera się do odbieralnika frakcję główną do momentu spadku temperatury.
Wyłączamy kolejno: pompkę wodną, płaszcz grzejny, po ostygnięciu roztworu wyrównuje się różnicę ciśnień wyjmując termometr albo kapilarę (należy to zrobić dość wolno).
Mierzy się objętość zebranego benzoesanu etylu, a także współczynnik refrakcji.
Opracowanie wyników
Kwas benzoesowy jest substancją stałą krystaliczną, temperatura topnienia białych kryształów wyznaczona wynosi 117 - 121 oC. Teoretyczna temperatura topnienia wynosi 122 oC, a więc różni się od wartości zmierzonej, co wskazuje na zanieczyszczenia. Wydajność wyodrębniania kwasu benzoesowego oraz krystalizacji, znając masę kwasu po krystalizacji - 1,35g:
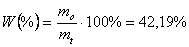
Przed destylacją objętość benzoesanu etylu wynosiła 20 cm3, a przeliczając na masę 20,84 g. Uzyskano 16,8 cm3 benzoesanu, czyli 17,51 g. Zatem wydajność procesu wynosi:
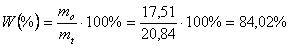
Chromatografia cienkowarstwowa i kolumnowa
Chromatografia służy do rozdziału kilku składników, opiera się na różnicy w wartościach współczynników podziału. W metodzie tej mamy do czynienia z dwiema fazami ruchomą: roztworem albo gazem oraz nieruchomą: ciało stałe albo ciecz, będąca w porach lub powierzchni substancji stałej. Faza ruchoma porusza się wzdłuż nieruchomej, a na ich granicy zachodzą zjawiska fizykochemiczne takie jak: adsorpcja chemiczna czy fizyczna, a także podział albo wymiana jonów. Różne składniki mieszaniny wykazują odmienne powinowactwo i dlatego następuje ich rozdział.
Rodzaje chromatografii:
- Bibułowa
- Kolumnowa
- Cienkowarstwowa
- Wstępująca
- Zstępująca
Chromatografia kolumnowa
W tej metodzie rozdział cieczy następuje między adsorbentem w postaci ciała stałego
a rozpuszczalnikiem. Jako adsorbenty stosuje się zazwyczaj: tlenek glinu (obojętny lub kwaśny lub zasadowy) oraz żel krzemionkowy, odpowiednio dobrany. Substancje stałe upycha się w szklanej pionowej kolumnie do jej połowy. Największą wydajność rozdziału można uzyskać, gdy stosunek długości kolumny do średnicy jest jak największy. Jednak w praktyce taki stosunek jest równy 10:1 do 20:1 ze względu na to, że przy większych wartościach duży przyczynek ma opór hydrauliczny. Aby rozdzielić 1g substancji ciekłej należy użyć 20 - 50 g adsorbenta. Na dole szklanej kolumny znajduje się kran bardzo szczelny, ale nie można używać smaru, ze względu na możliwość zanieczyszczenia wycieku.
Przy formowaniu kolumny najpierw umieszcza się watę, potem czysty piasek, dopiero później adsorbent stały, który uprzednio przechowuje się w roztworze o określonym składzie (pęcznieje), nie można wsypywać sproszkowanego adsorbenta (ze względu na nierównomierne rozmieszczenie i, co za tym idzie, mało skuteczny rozdział). Napełnianie musi się odbywać w umiejętny sposób, aby nie było pęcherzyków powietrza.
Formowanie kolumny
Rozpuszczalnik nalewa się do kolumny do 2/3 wysokości, następnie czopuje watą dół rurki. Spuszcza się tyle rozpuszczalnika, by w kolumnie nie zostały pęcherzyki powietrza (w razie konieczności należy uzupełnić braki). Potem wsypuje się powoli adsorbent. Drugim, znacznie częściej, stosowanym sposobem jest dodawanie do kolumny chromatograficznej adsorbenta pęczniejącego w rozpuszczalniku. Ważne jest, aby podczas spuszczania rozpuszczalnika dbać o to, by adsorbent zawsze był zwilżony przez rozpuszczalnik. Ciecz wlewa się z powrotem do kolumny. Osadzony adsorbent należy ostukać gumowym wężem, żeby przestał on osiadać. Na szczyt kolumny wsypuje się piasek, a potem bibułę (w postaci krążka). Takie działania zabezpieczają adsorbent przed unoszeniem podczas pracy kolumny.
Doboru mieszaniny rozpuszczalników oraz adsorbenta dokonuje się przy użyciu chromatografii cienkowarstwowej. Wykonuje się próbę, która sprawdza możliwość podziału mieszaniny.
Dobór eluenta oraz adsorbenta zależy od substancji, które mają być rozdzielone:
- Wymywanie składnika związanego z polarnym adsorbentem jest łatwiejsze w przypadku bardziej polarnego rozpuszczalnika.
- Im mieszanina jest bardziej polarna, tym bardziej niepolarny należy zastosować adstorbent oraz bardziej polarny eluent.
- W przypadku rozdziału cieczy o różnej mocy polarności, najpierw kolumnę przemywa się eluentem mniej polarnym, potem o coraz większej polarności, ale konieczne jest stopniowe zwiększanie polarnego charakteru rozpuszczalnika (w przeciwnym wypadku pasma zleją się ze sobą).
Rozdział substancji
Mieszaninę cieczy wprowadza się na szczyt kolumny w postaci roztworu (korzystna jest jak najmniejsza jego ilość oraz gdy jest to taki sam rozpuszczalnik, jaki zawiera kolumna) albo zawiesiny. Następnie należy spuścić powoli rozpuszczalnik do poziomu adsorbenta, tak by nie zapowietrzyć kolumny. Na szczyt kolumny wlewa się eluent, a więc następuje rozwijanie chromatogramu. Bardzo ważna jest kontrola wycieku roztworu z kolumny, na początku szybkość nie może przekraczać jednej kropli na sekundę, potem jej wartość może osiągnąć wartość do paru kropli w ciągu jednej sekundy. Można zaobserwować rozwarstwianie się rozdzielanej cieczy na tyle pasm ile jest składników. Kolejne porcje eluenta powodują wyciek poszczególnych substancji, które zbiera się
w oddzielnych odbieralnikach.
Czasem zdarza się, że rozdzielane ciecze nie zabarwiają chromatogramu, wówczas należy umieścić kolumnę
w lampie emitującej promieniowanie nadfioletowe. Innym sposobem jest uzyskanie informacji o frakcjach
z przeprowadzonej chromatografii cienkowarstwowej. Gdy jakieś frakcje mają taki sam skład należy je połączyć, gdy, natomiast, mamy do czynienia z międzyfrakcjami, czyli mieszaniną, to rozdziela się je ponownie, czasem odrzuca (gdy uzyskano zadowalającą ilość danej substancji). Związki stałe uzyskuje się przez odparowanie rozpuszczalnika na wyparce. Muszą one być poddane oczyszczeniu przy użyciu destylacji bądź krystalizacji.
Chromatografia cienkowarstwowa
W skrócie zwana także TLC (od angielskiego: thin layer chromatography), w której adsorbentem jest cienka warstwa (0,25 - 2,0 mm) takich substancji jak tlenek glinu czy żel krzemionkowy pokrywający szklaną płytkę albo folie (aluminiową, z sztucznego tworzywa). Ta technika służy do celów analitycznych lub preparatywnych, ponieważ umożliwia rozdzielenie nawet znikomych ilości substancji wynoszących 5 - 100 μg. Często metoda ta ma zastosowanie do wstępnego określania składu mieszanin.
Cechą charakterystyczną określonej substancji, przy zastosowanie określonego eluenta lub mieszaniny rozpuszczalników jest współczynnik RF , definiowany jako:
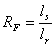
gdzie: ls - droga przebyta przez substancje rozdzielaną
lr - droga przebyta prze czoło rozpuszczalnika
Dla ściśle określonych warunków ciśnienia i temperatury współczynnik ten dla danego układu chromatograficznego ma stałą wartość.
Chromatogram musi być rozwijany w dość stabilnych warunkach, komorę stanowi szklany słoik albo zlewka ze szkiełkiem zegarkowym.
Metoda ta jest bardzo czuła i wymaga użycia małych ilości substancji, dlatego roztwory przeznaczone do rozdziału należy nanosić przy użyciu kapilar a odległość nie może być mniejsza niż 1 - 2 cm. Ważne jest, by plamki nanoszone miały małą średnicę, ponieważ wtedy uzyska się dokładniejszy rozdział. Oprócz plamek substancji, które maja być poddane rozdziałowi nanosi się również substancje wzorcowe. Płytkę umieszcza się
w komorze, do której uprzednio należy nalać ok. 0,5 cm eluenta, a następnie nakryć szkiełkiem zegarkowym. Po rozwinięciu chromatogramu (kiedy czoło rozpuszczalnika przebędzie drogę ok. 6 - 10cm), płytkę wyjmuje się, oblicza odległość punktu startowego od czoła rozpuszczalnika oraz suszy chromatogram.
Podobnie jak w przypadku chromatografii kolumnowej rozdział niektórych składników można zaobserwować nakierowując lampę ultrafioletową. Jednak w tym przypadku możliwe jest uzyskanie barwnego obrazu, widocznego dla ludzkiego oka, wywołując chromatogram przy użyciu odczynnika wywołującego, a więc takiego, który ulega reakcji z substancjami rozdzielonymi. Mogą nimi być ok. stężony kwas siarkowy (VI) czy pary jodu. W tym ostatnim przypadku do szczelnej komory wrzuca się parę kryształów jodu i oczekuje na utworzenie kompleksów o charakterystycznym zabarwieniu. Wiele ze związków organicznych tworzy z jodem substancje o specyficznej barwie, jedynie węglowodory nasycone oraz chlorowcopochodne nie posiadają takich własności.
Rozdział mieszaniny azobenzenu oraz o- nitroaniliny stosując chromatografie kolumnową, a kontrolująco - cienkowarstwową
Wykonanie ćwiczenia
Pod wyciągiem zestawia się sprzęt zgodnie z Rys. 6. Kolumna z kranikiem powinna mieć ok. 80 cm, a jej średnica - 1,5 - 2,5 cm. Eluentem jest roztwór heksanu (ewentualnie eteru naftowego) oraz octanu etylu
w stosunku 95:5. Jako adsorbenta należy użyć tlenku glinu (30 g). Kolumnę do rozdziału należy przygotować według wcześniej podanych instrukcji.

Rys. 6. Kolumna chromatograficzna
Substancje rozdzielane rozpuszcza się w niewielkiej ilości benzenu w probówce, potem wprowadza na szczyt kolumny. W fazie początkowej rozpuszczalniki wlewa się na szczyt kolumny. W chwili uwidocznienia pierścienia w dole kolumny zaczyna się zbierać poszczególne frakcje. Do pierwszej elenmajerki zbiera się frakcję "pomarańczową", potem nanosi na kolumnę mieszaninę heksan (eter naftowy) - octan etylu 70:30. Po zebraniu frakcji pośredniej, do trzeciej elenmajerki sączy się wyciek zawierający drugi składnik mieszaniny.
Metoda chromatografii cienkowarstwowej pozwala na określenie stopnia czystości rozdzielonych mieszanin,
także ich identyfikację.
Opracowanie wyników
Metoda chromatograficzna pozwoliła na rozdział azobenzenu oraz o-nitroaniliny. Potwierdzeniem otrzymania tych substancji jest przeprowadzenie chromatografii cienkowarstwowej. Związki były wystarczająco oczyszczone.
Zmierzono temperatury topnienia dla obu związków oraz porównano je z wartościami tablicowymi (Tabela 3.)
|
Związek
|
Wartość tablicowa
[oC]
|
Wartość doświadczalna
[oC]
|
|
azobenzen
|
68
|
63 - 68
|
|
o-nitroanilina
|
71,5
|
67 - 72
|
Tabela 3. Porównanie temperatur topnienia
Znając masy związków organicznych, które rozdzielano, wynoszących 0,3 g oraz zważonych po rozdziale: azobenzen - 0,28 oraz o-nitroanilina - 0,29 g można wyznaczyć wydajność procesu:
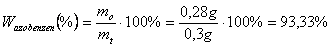
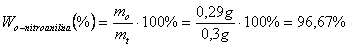
Wnioski
Chromatografia jest bardzo dobra metoda rozdzielania substancji zarówno pod względem czystości jak
i wydajności. Niestety jej wadami jest pracochłonność, czasem występuje problem z odpowiednim doborem eluenta, a także koszty związane z bardzo dużą czystością rozpuszczalników.
Synteza wybranych związków organicznych
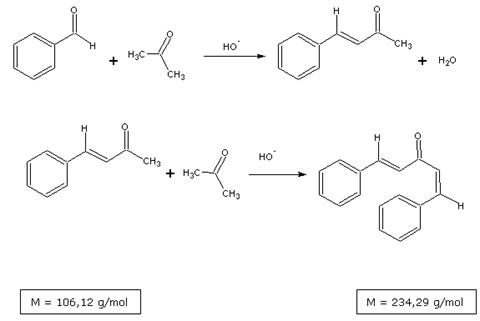
Dibenzylidenoaceton
Synteza dibenzylidenoacetonu należy do tak zwanej reakcji Claisena - Schmidta, charakteryzującej się kondesacją aldolową aldehydu o własnościach aromatycznych z aldehydem bądź ketonem alifatycznym. Produkt (aldol) ulega samorzutnej dehydratacji do związku karbonylowego nienasyconego. Wprowadzając do układu reakcyjnego benzaldehyd oraz aceton powoduje się powstanie benzylidenoacetonu (jako produktu pośredniego), a następnie dibenzylidenoacetonu w wyniku reakcji dwóch cząsteczek. Proces przedstawic można przy pomocy równania reakcji:
Wykonanie ćwiczenia
Do kolby okrągłodennej o objętości ok. 250 ml wsypuje się 7g wodorotlenku sodu, dodaje 70 ml wody destylowanej oraz 55 ml alkoholu etylowego. Przygotowaną mieszaninę stawia się na zimnej łaźni wodnej. Do kolby płaskodennej wlewa się 2,6 ml acetonu oraz 7,1 ml aldehydu benzoesowego wcześniej przedestylowanego. Do kolby okrągłodennej należy wlać ok. połowę objętości drugiej kolby i miesza się całość wykonując ruchy koliste. Po rozpuszczeniu składników należy włożyć kolbę do lodówki na ok. 0,5 h. Powstały osad musi być odsączony pod obniżonym ciśnieniem. Przemywanie woda destylowaną osadu prowadzi się do momentu uzyskania obojętnego odczynu, a potem odciska. W celu oczyszczenia otrzymanego związku należy poddać go krystalizacji z alkoholu etylowego.
Opracowanie wyników
Dibenzylidenoaceton to substancja stała, krystaliczna o żółtym zabarwieniu. Po krystalizacji zważono osad. Jego masa wynosiła 4,86 g. W celu obliczenia teoretycznej wartości masy produktu, przeliczono objętości użytych reagentów na mole:
Aldehyd benzoesowy: v = 7,1 ml (co stanowi 7,4053g, a zatem 0,0699 mola)
Aceton: v = 2,6 ml (a więc 2,0394g i 0,0352 mola)
Jak wynika z zapisu na jeden mol acetonu przypadają dwa mole benzaldehydu (użyto go w nadmiarze). Zatem, zakładając, że użyto 7,4053g aldehydu otrzymano by 8,17 dibenzylidenoacetonu (M = 234 g/mol). A skoro masa produktu wynosiła 4,86 g, więc można zapisać wyliczenie:
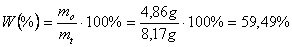
Zmierzono także temperaturę topnienia. Mieściła się ona w przedziale 104 - 109 oC. Porównując ta wartość
z tablicową: 112 oC, należy stwierdzić, że uzyskany produkt jest dość mocno zanieczyszczony.
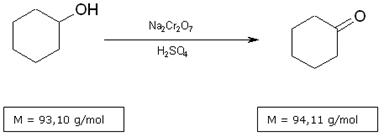
Cykloheksanon
Do otrzymywania ketonów używa się bardzo często alkoholi. Reakcji takiej ulegają wyłącznie drugorzędowe alkohole, ponieważ pierwszorzędowe zostają przekształcone w aldehydy, a trzeciorzędowe -
w ogóle nie reagują. Jako środki utleniające można zastosować: manganian (VII) potasu, tlenek manganu (IV) oraz związki chromu (VI).
Do reakcji cykloheksanolu użyto dichromianu sodu w kwaśnym środowisku. Wybór utleniacza jest powodowany szybkością oraz dużą wydajnością zachodzącej reakcji. Zachodzący proces można zobrazować następująco:
Duże znaczenie dla kierunku reakcji ma ścisła kontrola temperatury.
Wykonanie ćwiczenia
Do kolby płaskodennej o objętości ok. 250 ml dodaje się 60 g pokruszonego dość drobno lodu. Następnie wlewa się powoli 20 ml stężonego kwasu siarkowego (VI) pamiętając o ciągłym mieszaniu powstałego roztworu. Potem należy dodać 20,6 ml cykloheksanolu, całość ochłodzić na łaźni wodnej. W drugiej kolbie płaskodennej
o objętości 100 ml umieszcza się 25 g dichromianu sodu oraz wlewa 15 ml wody destylowanej. Powstały roztwór przelewa się do biurety. W pierwszej kolbie należy mierzyć temperaturę. Gdy wartość jej nie przekracza 15 oC dodaje się 1 ml roztworu dichromianu z biurety. Przy dodawaniu dichromianu następuje zmiana zabarwienia mieszaniny. Kolbę z cykloheksanolem należy, każdorazowo po dodaniu roztworu z biurety, mieszać. Kardynalną rzeczą jest, by temperatura nie była wyższa niż 45 oC. Najczęściej całkowity czas dodawania dichromianu sodu wynosi ok. 20 min. Wytworzony produkt poddaje się natychmiastowej destylacji. W tym celu przelewa się do kolby okrągłodennej o objętości 250ml poreakcyjną mieszaninę, zestawia aparaturę do destylacji, włącza płaszcz grzejny. Zbiera się ciecz, gdy temperatura mieszaniny osiągnie ok. 100 oC, a po uzyskaniu 10 - 15 ml destylatu zakończa się destylację. Wydzieloną ciecz wlewa się do rozdzielacza, następnie wsypuje 15 g chlorku sodu. Po starannym wytrząśnięciu (i rozpuszczeniu osadu) należy oddzielić frakcje cykloheksanonu. Roztwór suszy się nad bezwodnym siarczanem (VI) magnezu prze 15 - 20 min. Mieszaninę sączy się do kolby okrągłodennej (50 ml) prze sączek karbowany, montuje aparaturę destylacyjną. Zbiera się frakcję o temperaturze wrzenia 146 - 152 oC. Uzyskuje się miej więcej 12 g cykloheksanonu.
Opracowanie wyników
Cykloheksanon to ciecz o dość wysokiej temperaturze wrzenia. Z tego powodu destylację należało przeprowadzić z parą wodną. Po destylacji zmierzono objętość produktu. Wynosiła ona 5,1 ml, przeliczając na masę 4,52 g. Do układu reakcyjnego wprowadzono 20,3 ml cykloheksanolu, a więc 19,65 g. Zatem wydajność reakcji jest równa:
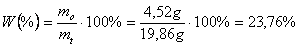
Zmierzono współczynnik refrakcji dla cieczy. Wartość doświadczalna 1,4489 nie pokrywa się z tablicową - 1,4513, co wskazuje na znaczne zanieczyszczenie substancji.
Wnioski
Przeprowadzona reakcja miała małą wydajność, co wynika z faktu wielu rozdziałów: destylacji, ekstrakcji. Wartość współczynnika załamania światła wskazuje na niewystarczającym osuszeniu produktu reakcji.
Chlorek tert-butylu
Chlorek tert-butylu zsyntetyzować można z alkoholu tert-butylowego dodając do układu reakcyjnego związki zawierające chlor takie jak: PCl5, chlorowodór, chlorek tionylu. Proces sprowadza się do substytucji grupy wodorotlenowej atomem chloru. W przypadku zastosowania kwasu chlorowodorowego reakcja opiera się na mechanizmie podstawienia nukleofilowego. Reakcja alkoholi trzeciorzędowych z kwasem solnym przebiega dość łatwo, podczas gdy alkohole drugorzędowe reagują dużo wolniej, a pierwszorzędowe ulegają temu procesowi bardzo trudno.
Reakcja przebiega według poniższego równania reakcji:
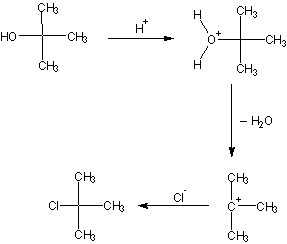
Wykonanie ćwiczenia
Do rozdzielacza o objętości 250 ml wlewa się 32 ml alkoholu tert-butylowego, 85 ml stężonego kwasu siarkowego (VI) i 10 g chlorku wapnia (bezwodnego). Ostatni składnik powoduje wzrost gęstości fazy kwasowej, a tym samym polepsza proces rozdziału. Mieszaninę należy wstrząsnąć kilka razy w czasie 20 min. W celu wyrównania ciśnienia po każdym wymieszaniu odtyka się korek. Po ustaleniu się równowagi, co obserwuje się "rozwarstwieniem" roztworu, spuszcza się dolną frakcję (kwaśną), po czym odrzuca (Rys. 7).

Rys.7. Rozdzielacz
Do górnej warstwy chlorku tert-butylu dolewa się 20 ml 5% roztworu wodnego wodorowęglanu sodu, w celu przemycia cieczy. Po przemyciu 20 ml wody destylowanej substancje przelewa się do kolby płaskodennej
o objętości 100 ml, a następnie suszy nad bezwodnym siarczanem (VI) magnezu (albo mad bezwodnym chlorkiem wapnia). Mieszaninę przesącza się przez sączek karbowany do okrągłodennej kolby ( v = 100ml). Po wrzuceniu kamyczka wrzennego przeprowadza się proces destylacji, stosując kolumnę Vigreaux. Frakcje zbiera się, kiedy temperatura wrzenia waha się w granicach 49 - 51 oC.
Opracowanie wyników
Chlorek tert-butylu to bezbarwna ciecz. Wyznaczając wydajność uwzględniono, że objętość użytego alkoholu wynosiła 32 ml, czyli 0,3378 mola. Z równania reakcji wynika, iż powstaje identyczna ilość moli,
a w przeliczeniu na gramy wynosi - 29,23g. W niniejszym doświadczeniu uzyskano 9,7 ml, co stanowi 7,67 g. Wydajność tego procesu wynosi, zatem:
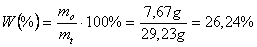
Porównanie wartości współczynników załamania światła: tablicowej 1,3856 oraz doświadczalnej - 1,3826 wskazuje na niezbyt staranne oczyszczenie substancji.
Wnioski
Chlorek tert-butylu uzyskano z bardzo małą wydajnością. Dość duży wpływ na taki stan miały straty w czasie ekstrakcji. Być może warstwę spuszczono, gdy równowaga nie ustaliła się dostatecznie. Ponadto na wydajność ma wpływ również przeprowadzona destylacja. Jak już podkreślono, współczynnik załamania doświadczalny wskazuje na dość duże zanieczyszczenie cieczy.
Mrówczan etylu
Jako typowy ester powstaje podczas reakcji estryfikacji, czyli procesu przebiegającego pomiędzy kwasami karboksylowymi a alkoholami. W przypadku tego estru, jako substratów używa się kwasu mrówkowego oraz etanolu, a reakcja przebiega zgodnie z równaniem:
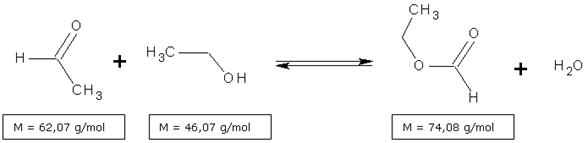
Reakcja przebiega w podwyższonej temperaturze, dodanie kwasu o dużej mocy (mrówkowego) znacznie wpływa na szybkość tworzenia się produktu. Dodatek w toku preparatyki, chlorku wapnia ułatwia zachodzenie procesu, a ciągła destylacja z mieszaniny reakcyjnej ma na celu, zwiększenie szybkości oraz wydajności reakcji.
Wykonanie ćwiczenia
W okragłodennej kolbie (100 ml) umieścić 15 ml 85% kwasu mrówkowego, 19 ml absolutnego (bezwodnego) etanolu oraz wsypać 3,7 g uwodnionego chlorku wapnia (CaCl2 * H2O). Naczynie z mieszaniną umieścić w zestawie destylacyjnym. W skład aparatury wchodzi również: kolumna Vigreux, termometr
(w nasadce destylacyjnej), chłodnica Liebiega, a także kolba płasko - albo okrągłodenna (o objętości 100 ml), jako odbieralnik, znajdującą się u wylotu chłodnicy w zimnej łaźni wodnej (chłodzenie). Mieszaninę reakcyjną ogrzewa się bardzo powoli do wrzenia (ok. pól godziny). Kiedy zakres temperatury ustali się
w przedziale 53 - 55 oC należy zbierać frakcję. Orientacyjny czas wycieku destylatu to 1godzina.
Powstający mrówczan etylu w ilości 25 ml (23g) jest zanieczyszczony. Dlatego też należy go poddać ponownej destylacji, której schemat został przedstawiony na Rys. 8.

Rys. 8. Aparatura służąca do destylacji prostej
Do układu należy użyć kolby okrągłodennej o objętości 100 ml. Do surowego estru dodaje się 3,3 g bezwodnego węglanu potasu, a także parę kamyczków wrzennych. Mieszaninę ogrzewa się w łaźni wodnej. Główny destylat zbiera się w przedziale temperaturowym 53 - 54 oC. Otrzymuje się orientacyjnie 22 g estru.
Opracowanie wyników
Mrówczan etylu ma postać bezbarwnej cieczy. Biorąc pod uwagę stechiometrię reakcji teoretyczna ilość estru jaka powinna powstać równa się liczbie moli kwasu mrówkowego - 0,3111 mola, przeliczając na gramy - 23,05 g. W wyniku reakcji powstało, natomiast, 20,5 ml mrówczanu, czyli 18,99 g. Wydajność procesu wynosi zatem:
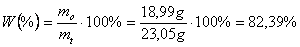
Pomiar współczynnika refrakcji mrówczanu etylu wykazał zgodność z tablicową wartością równą 1,3597.
Wnioski
Bardzo duża zbieżność współczynnika załamania światła jest charakterystyczna dla doskonale oczyszczonych substancji. Duża wydajność reakcji pozwala wnioskować, że metoda takiej syntezy jest wysoce skuteczna oraz powszechnie używana.
N-acetylo-p-toluidyna
Związki acylowe powstają w wyniku reakcji acylowania, których mechanizm opiera się na substytucji nukleofilowej. Najczęściej stosowane w syntezie organicznej są reakcje benzoilowania oraz acylowania.
Do tego typu procesów, jako czynnika acylującego, używa się zazwyczaj chlorków kwasowych, kwasów karboksylowych, bezwodników kwasowych, dużo rzadziej - pochodnych kwasów karboksylowych.
W przypadku N-acetylo-p-toluidyny stosuje się bezwodnik octowy, reakcja zachodzi według poniższego równania:
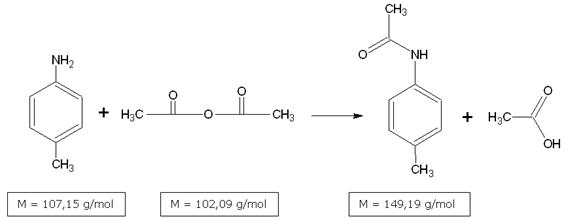
Wykonanie ćwiczenia
Do okrągłodennej kolby o objętości 100 ml wsypuje się 5,4 g p-toluidyny i wlewa 12,5 ml toluenu. Mieszaninę ogrzewa się na płaszczu grzejnym do rozpuszczenia używając chłodnicy zwrotnej. Roztwór odstawia się od źródła ogrzewania. Przez umieszczony wkraplacz w kolbie dodaje się porcjami bezwodnik octowy, utrzymując temperaturę poniżej 85 - 90 oC. Od 15 do 20 min należy utrzymać mieszaninę w tym zakresie temperatur. Po ostudzeniu wytrąca się krystaliczny osad p-acetylotoluidyny. Następnie odsącza się go oraz przemywa toluenem (kilka ml), potem suszy. Jeśli zachodzi taka konieczność to należy ponownie wykrystalizować osad z etanolu.
Opracowanie wyników
p-acetylotoluidyna jest substancja stałą, krystaliczną o żółtej barwie. Do reakcji zużyto 5,23 g
p-toluidyny, co stanowi 0,0489 mola. Teoretycznie masa powstałej p-acetylotoluidyny powinna wynosić 7,28 g,
a doświadczalna miała wartość 4,66 g. Na tej podstawie wydajność reakcji wynosi:
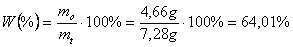
Określono także temperaturę topnienia tej substancji jako 146 - 153 oC. Porównując z wartością teoretyczną - 153 oC, można wnioskować o dość dobrym oczyszczeniu tego związku.
p-acetylotoluidyna nie ulega reakcji z typowymi zasadami, np. wodorotlenkiem sodu, ponieważ będąc sama amidem charakteryzuje się własnościami alkalicznymi.
Reaguje ona, natomiast, z kwasami, ale w bardzo nieznacznym stopniu, co świadczy o słabym charakterze zasadowym.
Wnioski
Nieduży rozrzut temperatury topnienia wskazuje na brak większego stężenia zanieczyszczeń. Mała wydajność to efekt, przede wszystkim krystalizacji, a także być może, niecałkowitego strącenia produktu z roztworu.
p-nitroacetanilid
Związki nitrowe powstają na wskutek reakcji nitrowania, w której ważnym składnikiem jest mieszanina nitrująca, czyli mieszanina stężonych kwasów: siarkowego (VI) oraz azotowego (V). W wyniku reakcji tych dwóch kwasów powstaje kation nitroniowy:
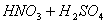

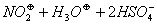
Atakuje on pierścień aromatyczny, powodując podstawienie elektrofilowe w pierścieniu aromatycznym (zastąpienie atomu H grupą NO2-).
Synteza p-nitoacetamidu opiera się na działaniu na acetanilid mieszaniną nitrującą:
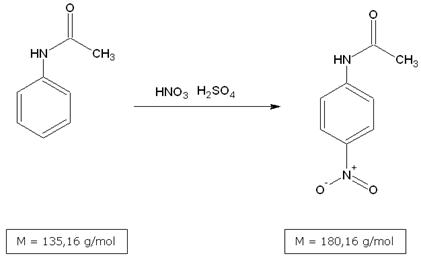
Wykonanie ćwiczenia
Do zlewki o objętości 100 ml wsypuje się 3,35 g suchego, roztartego w moździerzu acetanilidu, wlewa 3,4 ml kwasu octowego lodowatego. Do zlewki wrzuca się magnes i umieszcza na mieszadle magnetycznym. Stale mieszając wlewa się powoli 6,8 ml kwasu siarkowego (VI). Po rozpuszczeniu acetanilidu zlewkę wstawia się do naczynia zawierającego lód z solą. Przy użyciu wkraplacza dodaje się schłodzony roztwór stężonych kwasów azotowego (V) oraz siarkowego (VI) wmieszanych w stosunku 1,5 ml : 2 ml, kiedy roztwór w zlewce schłodzi się do temperatury 0 - 2 oC. Temperatura w zlewce nie może być wyższa niż 10 oC. Po wkropieniu mieszaniny kwasów zlewkę pozostawia się w pokojowej temperaturze prze okres ok. 0,5 godziny. Mieszaninę ze zlewki wlewa się do lodu pokruszonego (30 g) albo do 60 ml zimnej wody. Następuje wtedy krystalizacja p-nitroacetanilidu. Odczekać ok. 15 min., potem odsączyć kryształy na lejku Buchnera pod zmniejszonym ciśnieniem. Do przemywania używa się zimnej wody, koniecznie należy wymyć kwasy, co można sprawdzić wskaźnikami kwasowo - zasadowymi. Po odciśnięciu oraz osuszeniu p-nitroacetanilid krystalizuje się z 100 ml etanolu. Roztwór ochładza się, sączy, potem suszy w zakresie 60 - 80 oC. Przeciętnie otrzymuje się 2,5 g produktu.
Przed oczyszczaniem pobiera się małą ilość osadu do chromatografii.
Opracowanie wyników
p-nitroacetanilid jest bezbarwnym ciałem stałym, krystalicznym. Masa użytego acetanilidu wynosiła 3,32 g, co stanowi 0,024635 mola. Teoretycznie rzecz ujmując powinno powstać 4,43 g p-nitroacetanilidu (ze względu na stechiometrię reakcji). Kryształy produktu ważyły 3,64 g, tak, więc wydajność syntezy będzie równa:
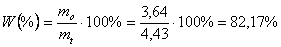
Określona została także temperatura topnienia p-nitroactanilidu jako 211 - 216 oC. Podobne wartości można spotkać w literaturze: 215 - 216 oC.
Wykonano także rozdział chromatograficzny p-mitroacetamidu metodą chromatografii cienkowarstwowej. Współczynnik RF jest równy:
RF =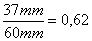
Wnioski
Chromatografia cienkowarstwowa pozwoliła na wysnucie wnioski, że otrzymany produkt nie zawiera znaczącej ilości zanieczyszczeń, ponieważ nie nastąpiło rozdzielnie na kilka plamek. Hipotezę tą potwierdza zbadana temperatura topnienia, której wartość pokrywa się z literaturą.
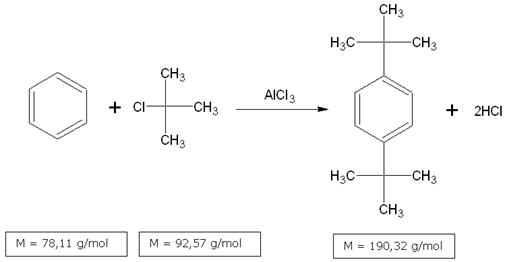
1,4 - di-tert- butylobenzen
Synteza tego związku organicznego opiera się na reakcji Friedla - Craftsa, której sednem jest alkilowanie alko acylowanie związku o charakterze aromatycznym halogenkiem alkilu bądź acylu w środowisku AlCl3. Chlorek glinu powoduje powstanie karbokationu, który będąc czynnikiem elektrofilowym atakuje pierścień. 1,4-di-tert-butylobenzen powstaje stopniowo: najpierw tworzy się monopochodna, dopiero później -produkt z dwoma podstawionymi grupami. W wyniku takiego rodzaju reakcji tworzy się mieszanina dwóch izomerów: orto i para. Proces ten można zapisać równaniem:
Ważne jest zastosowanie suchego chlorku glinu. Należy do przechowywać np. w eksykatorze, ze względu na bardzo dużą higroskopijność. W przypadku powstania grudek można rozbić je w moździerzu. Podobnie jak omawiany odczynnik, również inne substancje chemiczne oraz stosowany sprzęt musi być suchy. Syntezę należy przeprowadzać pod dygestorium, ponieważ podczas tego procesu wydziela się chlorowodór, a benzen jest silnie toksyczny.
Wykonanie ćwiczenia
Do osuszonej okragłodennej kolby (o objętości 100 ml) wlewa się 11,5 ml chlorku tert-butylu i 4,4 ml benzenu. Wkłada rurkę zawierającą bezwodny chlorek wapnia. W kolbie płaskodennej odważa się 0,5 g bezwodnego chlorku glinu. Do kolby reakcyjnej wsypuje się 1/8 całkowitej ilości chlorku glinu. Dodanie każdej porcji tego związku powoduje wydzielanie chlorowodoru. Mieszaninę należy mieszać kolistym ruchem kolby. Po zaprzestaniu wydzielania gazu wsypuje się kolejną porcję chlorku i tak do skrzepnięcia mieszaniny. Jeśli ten proces nie następuje (powinien nastąpić w ciągu 20 min.) to dodaje się tyle AlCl3, by uzyskać stałą konsystencję. Następnie wlewa się 25 ml eteru etylowego, powoli wkrapla 10 ml wody destylowanej, mieszając zawartość kolbki. Powstaje bezbarwny roztwór, który należy przelać do rozdzielacza. Spuszcza się warstwę wodną
i odrzuca, warstwa eterowa zostaje przemyta 10 ml wody destylowanej. Oddziela się frakcje, do roztworu eterowego dodaje się bezwodnego siarczanu (VI) sodu albo magnezu (1 g) w celu osuszenia. Mieszaninę sączy się, przemywając 5 - 10 ml eteru do okrągłodennej kolby. Roztwór destyluje się, podgrzewając na łaźni wodnej, a jako odbieralnik używa się cylindra miarowego. Gdy wycieknie 15 ml destylatu, destylację przerywa się,
a kolbę schładza. Można wspomóc krystalizacje przez pocieranie ścianek bagietką. Kryształy sączy się na lejku Schotta, odciska, suszy na powietrzu. Zazwyczaj uzyskuje się 7g 1,4-di-tert-butylobenzenu.
Opracowanie wyników
Produkt jest bezbarwnym ciałem stałym o strukturze krystalicznej. Na podstawie ilości użytych substratów: 4,4 ml (3,84 g, 0,0492162 mola) benzenu i 11,4 ml (9,6 g, 0,1036923 mola) chlorku tert-butylu, wyznaczono teoretyczną masę 1,4-di-tert-butylobenzenu, jako 9,37 g. W rzeczywistości uzyskano 6,85 g. Zatem wydajność syntezy przedstawia się równaniem:
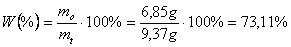
Określono również temperaturę topnienia w przedziale 76 - 79 oC, podczas gdy wartość teoretyczna wynosi
78 oC.
Wnioski
Podobieństwo temperatur topnienia rzeczywistej oraz literaturowej świadczy o dobrze oczyszczonym związku.
