Wstęp:
Chiralność to cecha wielu związków naturalnych. Chiralnością odznaczają się zarówno cząsteczki DNA, ale także cząsteczki enzymów, hormonów oraz feromonów. Dla przykładu enancjomery limonenu różnią się zapachem. (R)-limonen ma zapach pomarańczy, natomiast (S)-limonen cytryn.
Warunek chiralności:
Podstawowym warunkiem chiralności jest to, aby cząsteczka miała atom węgla, który łączy się 4 różnymi podstawnikami
(tzw. centrum stereogeniczne, centrum chiralności). Istnieją wówczas dwa układy różniące się strukturą w przestrzeni. Układy te maja takie same identyczne połączenia atomów w cząsteczce (konstytucje). Układy nazywamy enancjomerami, jeżeli porównując je do siebie możemy stwierdzić, że mamy do czynienia z obiektem i jego lustrzanym odbiciem. Gdy w cząsteczce jest większa ilość atomów węgla, to możemy utworzyć więcej struktur w przestrzeni, o takim samym połączeniu atomów w cząsteczce. Są to stereoizomery, które dzielą się na enancjomery i diastereoizomery. Ogólny wzór na liczbę możliwych stereoizomerów wynosi 2n, gdzie chóralnych to liczba optycznie czynnych atomów, które są połączone z 4 różnymi podstawnikami.
Związki chiralne to związki optycznie czynne. Związki te potrafią skręcać płaszczyznę światła spolaryzowanego. Najliczniejsza grupę substancji zdolnych to tego, to związki, posiadające swojej cząsteczce atom lub atomy węgla, które są połączone z 4 podstawnikami różniącymi się między sobą. Czynność optyczna jest możliwa wtedy, kiedy roztwór zawiera jeden enancjomer w większej ilości. Mieszanina zawierające równe ilości enancjomerów nie skręca płaszczyzny spolaryzowanego światła. Czyste enancjomery mogą skręcać płaszczyznę światła pod takim samym kątem, jeśli chodzi o wartość bezwzględną, ale przeciwną, jeśli chodzi o znak.
Jak cząsteczki związków optycznie czynnych powodują skręcenie płaszczyzny światła spolaryzowanego
Promień spolaryzowanego światła w płaszczyźnie to suma wektorowa 2 promieni, które ulegają polaryzacji kołowej w przeciwnych kierunkach. Promienie w inny sposób oddziałuje z elektronami walencyjnymi badanego związku. Dochodzimy do wniosku, że składowe promienia kołowo spolaryzowanego, gdy przechodzą przez badany roztwór zawierający związek chiralny zupełnie inaczej z nim oddziałują. Płaszczyzna wektora wypadkowego musi ulec skręceniu. Substancję nazywamy prawoskrętna, jeżeli płaszczyzna ulega skręceniu zgodnie ze wskazówkami zegara, wówczas do nazwy związku dodajemy znak (+). Natomiast substancja lewoskrętna, to taka, której płaszczyzna ulega skręceniu niezgodnie ze wskazówkami zegara, wówczas do nazwy związku dodajemy (-).
Określanie konfiguracji związku chiralnego
Za przykład przysłuży nam cząsteczka alaniny, która zawiera 1 atom węgla, połączony z czterema niejednakowymi podstawnikami. Występują zatem dwa enancjomery.
Wzór strukturalny alaniny wygląda następująco:

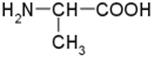
Zgodnie z panującą zasadą wiązania poziome wychodzą z płaszczyzny rysunku w stronę obserwatora, natomiast wiązania pionowe „ukrywają się” pod płaszczyzną rysunku.
Jak określić konfigurację? Na początku musimy ustalić sekwencję czterech podstawników, różniących się od siebie przy centralnym węglu. Zasada obowiązujące w tym przypadku to liczba atomowa pierwiastka połączonego bezpośrednio z asymetrycznym węglem. Pierwiastki o większej liczbie atomowej są ważniejsze. W cząsteczce alaniny kolejność jest następująca: N, C, H. Azot charakteryzuje się największą liczbę atomową, dlatego podstawnikiem najważniejszym może być tylko grupa -NH2, zaś wodór o liczbie atomowej najmniejszej, jest podstawnikiem najmniej ważnym. Chcąc ustalić kolejności podstawników -CH3 oraz –COOH musimy wziąć pod uwagę atomy, z którymi bezpośrednio są połączone atomy węgla i jeszcze raz zastosować zasadę liczby atomowej. Gdy wykonamy to zadanie, to jesteśmy w stanie ułożyć podstawniki pod katem ważności. W przypadku alaniny mamy: -NH2 później -COOH, -CH3 i na końcu -H. Kolejnym zadaniem, przed którym stoimy, to ustawienie cząsteczki tak, aby spoglądać na H przez atom węgla. Trzy pierwsze podstawniki kierują się stronę patrzącego, a atomu H (jako najlżejszy podstawnik) nie widać.
Przy takim ustawieniu cząsteczki następnym etapem będzie ustalenie kierunku, w którym ulega zmniejszaniu ważność podstawników. Gdy zmiana jest zgodna ze wskazówkami zegara, to mamy do czynienia z konfiguracją R (od łac. rectus = prawy), jeśli zmiana jest odwrotna, to mamy do czynienia z konfiguracją S (od łac. sinister = lewy). Konfiguracja określona w ten sposób jest bezwzględna, gdyż pod uwagę wzięliśmy tylko masy atomów.
W przypadku alaniny otrzymamy cząsteczki: (R)-alanina i (S)-alanina. Jeśli w nazwie dodatkowo chcemy umieścić informacje o kierunku skręcania płaszczyzny spolaryzowanego światła, to otrzymamy następujące cząsteczki: R)-(-)-alanina i (S)-(+)-alanina. Warto podkreślić, że kierunek zmniejszającej się ważności podstawników nie pokrywa się z stroną skręcania płaszczyzny spolaryzowanego światła.
W praktyce dla cukrów oraz aminokwasów posługujemy się tzw. konfiguracjami względnymi. Określane są one przez porównanie badanej cząsteczki do struktury aldehydu D-(+)glicerynowego.
Wzór aldehydu D-(+)glicerynowego wygląda następująco:
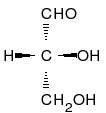
Chcą porównać badaną cząsteczkę do zaprezentowanej powyżej musimy ją przedstawić w tzw. projekcji Fischera. Pionowo musi być ustawiony najdłuższy łańcuch, zaś pierwszy węgiel musi znajdować się na samym szczycie. Dla aldehydu glicerynowego interesuje nas konfiguracja przy węglu C2. Zauważamy, że grupa -OH leży po prawej stronie, mamy do czynienia zatem z konfigurację D, określoną mianem konfiguracji naturalnych cukrów prostych. W przeciwnym przypadku, gdy grupa OH leży po lewej stronie, to mamy do czynienia z konfigurację L tzw. konfiguracja naturalnych aminokwasów.
Reakcje związków chiralnych
W przypadku związków chiralnych mamy do czynienia z reakcjami selektywnymi. Są to takie reakcje, w których z kilku możliwych do uzyskania produktów w większej ilości powstaje jeden. Jeżeli pod wpływem reakcji uzyskujemy nowe centrum chiralne, to możliwe jest zarówno konfiguracja R oraz S. Z enancjoselektywnością mamy do czynienia wtedy, gdy pod wpływem reakcji tworzą się enancjomery, gdy powstają diastereoizomery, to mamy do czynienia z diastereoselektywności. Skrót ee wyraża wzajemne proporcje stereoziomerów ( nadmiar enancjomerów) , a skrót de (nadmiar diastereoizomerów).
Jak obliczamy ten nadmiar? Jest to iloraz różnicy zawartości obydwu form przez ich wzajemną sumę:
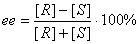
R oraz S to powstałe enancjomery;
[R] oraz [S] to ułamki molowe, ale [R] > [S]
Z powyższego równania można obliczyć nadmiar enancjomerów wyrażony w procentach.
Katalizatory używany w reakcjach związków chiralnych
Czynnikiem indukcyjnym wprowadzany do reakcji, niezbędnym do utworzenia centrum stereogenicznego, była substancja o znanej budowie przestrzennej. Żeby uzyskać dużą liczbę diastereoizomerów wprowadzano do cząsteczek pomocniki chiralne, których zadaniem było blokowanie dostępu do reagującej części z jednej strony. Stosowane katalizatory miały istotny wpływ na selektywność zachodzącej reakcji, ale nie były związkami chóralnymi.
Ostatnimi czasy radykalnie zmieniło się myślenie o syntezie związków chiralnych. Zgodnie z nową tendencją nośnikiem niezbędnych informacji o strukturze oraz wzajemnym stosunku obu enancjomerów jest katalizator. Jest to substancja, która nie bierze udziału w reakcji i jest używana w małej ilości około 0,5%. Mamy do czynienia z katalizą homogeniczną, tzn. wspomniane katalizatory ulęgają rozpuszczeniu w rozpuszczalnikach organicznych. Nie mogą tworzyć odrębnej fazy. Katalizator jest optycznie czystym związkiem chiralnym. Katalizatory, które są używane w reakcji redukcji są kompleksami jonów metali grup przejściowych (Rh, Ru), z optycznie czynnymi ligandami pochodzenia organicznego.
Mechanizm katalitycznej redukcji
Reakcja ta polega na utworzeniu przez jon metalu wiązań kompleksowych, wchodzący w skład katalizatora, z parami elektronowymi tlenu oraz azotu, ewentualnie, wiązaniami substratów, Przyłączanie wodoru do substratu zachodzi z jednej strony łatwiej niż z drugiej. Uzyskujemy w taki sposób znaczne ilości określonych enancjomerów.
Asymetryczne reakcje redukcji przeprowadzone wobec chóralnych katalizatorów umożliwiają otrzymać oczekiwany, czysty optycznie enancjomer ze znaczną wydajnością.
Przy pomocy tej metody jesteśmy w stanie obniżyć koszty produkowania związków chóralnych. Jest to niesłychanie ważne w reakcjach syntezy substancji biologicznie czynnych, do których zaliczamy leki i środki ochrony roślin.
Przykłady syntez związków chiralnych
Według nowej koncepcji pierwszym procesem, opracowanym przez zespół pod kierownictwem William S. Knowlesa, katalitycznego wodorowania na skale przemysłową jest reakcja syntezy związku podawanego chorym na Parkinsona. Był to związek o nazwie L-Dopa. Związek uzyskiwany w katalitycznym uwodornieniu jest czystym enancjomerem typu S.
Za pomocą tej samej metody otrzymano także (S)-fenyloalaninę, stosowaną w produkcji aspartamu.
Kontynuator prac Williama S. Knowlesa, japoński chemik, Ryōji Noyoriego używając kompleksu (S)-BINAP- Ru(OCOCH3)2 dokonał pomyślnie redukcję i otrzymał (S)-naproksen – lek działający przeciwzapalnie, przeciwgorączkowo oraz przeciwbólowo. Uzyskał niesamowitą wydajność reakcji 92%. Nadmiar enancjomerów (ee) wyniósł 97%.
Związki kompleksowe rutenu z BINAP-em są katalizatorami w asymetrycznej reakcji redukcji ketonów do alkoholi. Redukcji ulega wiązanie ketonowe C=O.
Z kolei drugi ze współpracowników Williama S. Knowlesa, amerykański chemik, Karl Barry Sharpless zajął się później reakcjami utleniania związków organicznych. Występuje w nich etap polegający na kompleksowaniu chiralnych ligandów jonem metalu przejściowego- w tym przypadku osmu (Os). Amerykański badacz jako ligand zastosował pochodną chininy. W procesie tym zastosowano:
- niewielkie ilości tlenku osmu(VIII)- użytego jako czynnik kompleksujący;
- małą ilość pochodnej chininy (ligano);
- nadmiar N-tlenku-4-metylomorfoliny (NMO).
Środowiskiem reakcji była mieszanina aceton-woda. Reakcję przeprowadzano w temperaturze 0°C, przez około 15 godzin. Uzyskano produkt z 89 procentową wydajnością.
