ELEKTROLIZA
1) Czym jest elektroliza?
Elektroliza to jeden z procesów elektrochemicznych, dzięki któremu można wywołać przemiany chemiczne przy użyciu energii elektrycznej. Nie dotyczy to wszystkich przemian - do przeprowadzenia elektrolizy niezbędne są swobodnie poruszające się jony. Można je znaleźć w stopionym elektrolicie lub w roztworze elektrolitu. Ważne jest, żeby rozpuszczalnik takiego roztworu był polarny (najczęściej korzysta się z wody).
2) Na czym polega?
Energia elektryczna wykorzystywana w procesie elektrolizy dostarczana jest w postaci prądu stałego. Jest on czerpany z źródła zewnętrznego (np. z prądnicy czy z akumulatora). Prąd doprowadza się do elektrolitu przez elektrody wykonane z metalu lub grafitu. Przyłożenie napięcia do elektrod wywołuje między nimi pole elektryczne. To z kolei sprawia, że aniony (ładunek ujemny) znajdujące się w elektrolicie kierują się w stronę anody - elektrody, do której przyłożono dodatni biegun źródła prądu. Kationy, o ładunku dodatnim, podążają w stronę katody - elektrody, do której przyłożono ujemny biegun źródła prądu. W pewnym momencie jony stykają się z odpowiednimi elektrodami i wymieniają z nimi elektrony. Zetknięcie się z katodą powoduje pobieranie elektronów, następuje reakcja redukcji. W efekcie jony stają się obojętnymi drobinami i w tej postaci wydzielają się na katodzie. Zetknięcie się z anodą powoduje oddanie elektronów, dochodzi do reakcji utleniania i wydzielenia obojętnych drobin na anodzie. Te zależności są prawdziwe zawsze - bez względu na substancję poddawaną elektrolizie. A więc na katodzie zawsze zachodzi reakcja redukcji, a na anodzie - utleniania.
3) Co daje? Jak można nią sterować?
Przeprowadzając elektrolizę można uzyskać różnorodne substancje, w zależności od warunków panujących podczas całego procesu. Oto możliwości zmiany warunków:
- substancją wyjściową jest stopiony elektrolit albo jego wodny roztwór
- roztwory mogą mieć różne stężenia
- można zmieniać napięcie przykładane do elektrod (np. poprzez wybór różnych źródeł prądu)
- można używać elektrod wykonanych z różnych materiałów (np. inny produkt można uzyskać na elektrodach miedzianych, a inny na grafitowych).
4) Jak ją opisać?
Faraday sformułował prawa pozwalające na ilościowe opisanie przemian zachodzących podczas elektrolizy. Jego prace nad prądem elektrycznym i jego właściwościami dostarczyły wiele wiadomości m.in. o jonach i elektrolizie (on jest twórcą pojęcia elektroliza). Przeprowadził serie doświadczeń mających na celu zbadanie procesu elektrolizy. Analiza danych i wnikliwe rozważania doprowadziły go do odkrycia pewnych prawidłowości. Sformułował je w postaci dwóch nieskomplikowanych praw zwanych dziś prawami Faradaya. Oto one:
1) Masa substancji wydzielonej przy elektrodzie nie zależy ani od właściwości roztworu, ani od charakteru elektrod i zmienia się wprost proporcjonalnie do wielkości ładunku elektrycznego przepuszczonego przez elektrolizer. Zależność ta opisana jest wzorem:
m = kq = kIt
m - masa substancji wydzielonej na elektrodzie
k - współczynnik proporcjonalności ("równoważnik elektrochemiczny, czyli współczynnik określający masę substancji wydzielonej przy przepływie jednostkowego naboju, np. jednego kulomba" - wg K.M. Pazdro "Podstawy chemii dla kandydatów na wyższe uczelnie")
q - wartość ładunku przeniesionego przez jony
I - natężenie prądu
t - czas trwania elektrolizy
2) "stosunek masy molowej substancji wydzielającej się na elektrodzie do iloczynu jej równoważnika elektrochemicznego i liczby ładunkowej reakcji elektrodowej (zapisanej dla jednego mola substancji o masie molowej M) jest wielkością stałą dla wszystkich procesów elektrodowych i wynosi 96 500 C/mol" (wg K.M. Pazdro "Podstawy chemii dla kandydatów na wyższe uczelnie"):
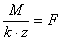
M - masa molowa substancji wydzielonej na elektrodzie
z - liczba ładunkowa
F - stała Faradaya = 96 500 C/mol
Oba prawa można połączyć i przedstawić w postaci jednego wzoru:
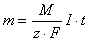
5) Jakie ma praktyczne zastosowania?
Elektroliza znalazła wiele zastosowań praktycznych, szczególnie w przemyśle ale również w analizie ilościowej. Procesy wykorzystywane w przemyśle można sklasyfikować następująco:
- Elektroliza roztworów wodnych, w tym:
a) procesy, podczas których metal nie wydziela się katodzie. Zaliczamy do nich m.in.: uzyskiwanie czystego H2 poprzez rozłożenie na pierwiastki wody, którą wcześniej zakwaszono albo zalkalizowano; otrzymanie chloranu lub podchlorynu sodowego (używanych jako mocne środki wybielające czy też utleniające) przez utlenienie chlorku sodu (popularnej soli kuchennej); otrzymywanie nadtlenku wodoru (H2O2) przez rozkład związków nadtlenowych otrzymanych przez utlenienie kwasu siarkowego(VI) albo siarczanu amonu [(NH4)2SO4].
b) procesy, w wyniku których metal wydziela się na katodzie. Wydzielenie metalu jest wtedy bardzo pożądane i stanowi wręcz cel przeprowadzenia elektrolizy. Można w ten sposób uzyskać metale o wysokiej czystości, gdyż wszelkie zanieczyszczenia w znakomitej większości pozostaną w roztworze. Stąd też korzysta się z tej metody przy rafinacji miedzi hutniczej. Zanieczyszczona miedź spełnia rolę anody w elektrolizerze a czysta miedź (zwana miedzią elektrolityczną) wydziela się na powierzchni katody. W ten sam sposób oczyszcza się cynk z rud cynku (proces zachodzi w kwasie). Inne zastosowanie tego typu elektrolizy to pokrywanie różnych powierzchni powłoką galwaniczną z metali szlachetniejszych. Otrzymane powłoki mogą mieć charakter dekoracyjny lub ochronny jak w przypadku niklowania czy chromowania stali. W podobny sposób otrzymuje się formy galwanoplastyczne. Można je uzyskać na modelach, przykładowo - gipsowych, które pokryte zostały warstwą grafitu (grafit sprawia, że nabierają zdolności do przewodzenia prądu, a więc mogą stanowić katodę)
- Elektroliza stopionych elektrolitów (inaczej - termoelektroliza). Ten rodzaj elektrolizy wykorzystuje się m.in. do uzyskiwania wolnego od zanieczyszczeń glinu z tlenku glinu rozpuszczonego w kriolicie. Jest to też dobra metoda otrzymywania metali lekkich (głównie sodu, wapnia i magnezu) ze stopionych soli tych metali.
6) Czym są elektrolizery?
Aby przeprowadzić elektrolizę trzeba posłużyć się specjalnymi aparatami zwanymi elektrolizerami. W przemyśle korzysta się z elektrolizerów w postaci otwartych lub zamkniętych zbiorników (różnego rodzaju kotły, wanny czy bębny) wypełnionych elektrolitem. Z elektrolitem mają styczność (np. przez zanurzenie) przynajmniej 2 elektrody (przykładowo grafitowa i metalowa), podłączone do źródła prądu stałego. W przypadku elektrolizera bezprzeponowego miejsc wokół katody i anody nie rozdziela się. Odwrotnie jest w przeponowym elektrolizerze - tam pomiędzy anodą a katodą umieszcza się przegrodę w postaci metalowej siatki pokrytej materiałem izolującym lub porowatej płytki ceramicznej albo przepony azbestowej. Taka przegroda umożliwia przepływ prądu jednocześnie zapobiegając wzajemnemu mieszaniu produktów powstałych na anodzie i katodzie (nie dochodzi do reakcji między nimi). Taką samą funkcję ma dzwon w elektrolizerach nazywanych dzwonowymi. Nakłada się go z góry na 1 lub 2 elektrody. Jest on nie tylko przegrodą ale także umożliwia odprowadzanie produktów gazowych elektrolizy. Termoelektrolizery to typ elektrolizerów przystosowanych do przeprowadzania elektrolizy substancji w fazie stałej. Z kolei galwanotechnika posługuje się tzw. galwanicznymi wannami.
6) Podsumowanie najważniejszych informacji
Połączenie elektrod metalicznych zanurzonych w roztworze pewnego elektrolitu (albo innego przewodnika o charakterze jonowym) z źródłem energii elektrycznej (musi to być źródło zewnętrzne prądu stałego) generującym odpowiednie napięcie, powoduje zajście procesów znanych pod nazwą elektrolizy. Elektroliza jest przeprowadzana w układzie noszącym nazwę elektrolizera. Dodatni biegun źródła prądu połączony jest z elektrodą zwaną anodą, natomiast ujemny biegun - z katodą. Anoda jest elektrodą, na której zawsze zachodzi utlenianie, a na katodzie zawsze zachodzi redukcja. Elektroliza może zajść tylko wtedy, gdy napięcie doprowadzone z zewnętrznego źródła energii elektrycznej będzie wyższe od napięcia analogicznego ogniwa (tzn. takiego, w którym zachodzą reakcje przeciwne do reakcji zachodzących w elektrolizerze).
To, co determinuje kierunek i rodzaj reakcji, jakie zajdą na obu elektrodach w trakcie przepływu ładunków przez układ elektrolizera, to głównie skład chemiczny roztworu, materiały, z których zbudowane są elektrody, oraz napięcie i temperatura układu. Nie ma jednoznacznych, skutecznych w każdym przypadku zasad przewidywania zachodzących procesów, chociaż istnieje kilka prostych reguł pozwalających przewidzieć wiele sytuacji. Najważniejsze żeby pamiętać, że na danym typie elektrody (dodatnia lub ujemna) zachodzi tylko jeden proces (redukcja lub utlenianie). Warto także wiedzieć, że w reakcji anodowej uczestniczą tylko drobiny, które mogą oddać elektrony (będzie to dla nich korzystne energetycznie) a w reakcji katodowej tylko te, które mogą pobrać elektrony.
Według definicji elektrolizy jej przebieg nie jest ograniczony tylko do środowiska wodnych roztworów elektrolitów. Może ona zachodzić także w przewodniku jonowym. Dotyczy to głównie stopionych substancji mających budowę jonową, do których zaliczamy np. tlenki metali i wodorotlenki. W wyniku topienia tych substancji dochodzi do zniszczenia ich regularnej budowy krystalicznej. Sieć krystaliczna rozluźnia się do tego stopnia, że jony mogą poruszać się swobodnie. To sprawia, że mogą brać udział w reakcjach zachodzących na elektrodach oraz mogą przenosić ładunki elektryczne (co jest równoznaczne z przewodzeniem prądu).
OGNIWA GALWANICZNE
Pewnie większość z nas nie zdaje sobie sprawy, że niemal codziennie posługuje się różnymi rodzajami ogniw. Służą one m.in. do zasilania magnetofonów, odbiorników radiowych, zegarków, kalkulatorów i tym podobnych urządzeń elektrycznych.Po raz pierwszy ogniwo skonstruowano około 200 lat temu. Dokonał tego fizyk włoski Alessandro Volt ( żył w latach 1745-1827 ). Dzisiejsze ogniwa mają nieco inną formę, przez te 200 lat przeszły wiele przeobrażeń, które doprowadziły je do współczesnej postaci.
Termin ogniwo galwaniczne wywodzi się od nazwiska pewnego włoskiego fizyka, fizjologa i lekarza Luigi Galvaniego, który żył w latach 1737-1798. Ogniwem galwanicznym nazywamy układ składający się z 2 półogniw (nie mogą być to te same półogniwa), który jednocześnie spełnia warunek, że w wyniku połączenia półogniw za pomocą zewnętrznego przewodnika metalicznego w układzie popłynie prąd. Przepływ elektronów pomiędzy półogniwami jest możliwy dzięki różnicy wewnętrznych potencjałów między obiema częściami układu. Półogniwa można łączyć w sposób bezpośredni lub z wykorzystaniem przegrody umożliwiającej przemieszczanie się jonów.
Półogniwem nazywamy układ co najmniej dwufazowy złożony z elektrody (faza metaliczna) i roztworu elektrolitu, w którym elektroda jest zanurzona. Znanych jest wiele typów półogniw, wśród nich możemy wyróżnić m.in. półogniwa typu redoks, gazowe czy metaliczne. W przypadku półogniw redoks elektroda służy tylko jako przenośnik elektronów, sama nie jest ani substratem ani produktem zachodzącej reakcji. Podane warunki dobrze spełniają platyna i grafit, dlatego też są to najczęściej stosowane substancje do budowy półogniw tego typu. Półogniwo gazowe to rodzaj półogniwa redoks. Składa się ono z blaszki metalu, który jest na tyle szlachetny, że nie uczestniczy w reakcji (najczęściej stosuje się platynę) i z roztworu, w którym elektroda jest zanurzona. Roztwór ten jest nasycony gazem, stale dostarczanym w trakcie pracy ogniwa i zawiera jony będące utlenionymi bądź zredukowanymi formami cząsteczek tego gazu. Do najpopularniejszych półogniw gazowych zaliczamy półogniwo chlorowe i wodorowe (ich połączenie tworzy układ, w którym można przeprowadzić syntezę chlorowodoru). Obok półogniw redoks popularne są także półogniwa metaliczne. W tym przypadku elektrolit, w którym zanurzona jest metalowa blaszka, zawiera jony tego samego metalu. Wobec tego elektroda nie jest już bierna ale uczestniczy w reakcji chemicznej zachodzącej w półogniwie.
Aby w szybki sposób zapisać układ ogniwa galwanicznego wprowadzono pewną konwencję tworzenia schematów ogniw bez konieczności rysowania ich:
Elektroda I | elektrolit I || elektrolit II | elektroda II
Inaczej można to zapisać w postaci :
(-) anoda | elektrolit anodowy || elektrolit katodowy | katoda (+)
Przykładowo:
(-) Zn | ZnSO4 || CuSO4 | Cu (+)
Znaki (+) i (-) przedstawiają bieguny ogniwa i wskazują kierunek przepływu prądu. Pionowa kreska oddziela symbol fazy metalicznej od symbolu odpowiadającego jej elektrolitu, a dwie pionowe kreski symbolizują obecność klucza elektrolitycznego.
Podany w powyższym przykładzie schemat opisuje budowę popularnego ogniwa Daniela, który składa się z dwóch oddzielonych układów - płytki cynkowej zanurzonej w roztworze siarczanu (VI) cynku (ZnSO4) i płytki miedzianej zanurzonej w roztworze siarczanu (VI) miedzi (II) (CuSO4). Te półogniwa najczęściej oddziela się od siebie kluczem elektrolitycznym, przez który mogą swobodnie migrować jony i wyrównywać tym samym poziom ładunków elektrostatycznych między roztworami.
Ogniwa używane na co dzień w mowie potocznej nazywane są bateriami. W ujęciu chemicznym bateria jest zestawem połączonych dwóch lub trzech ogniw (szeregowo lub równolegle). Popularne "paluszki" są pojedynczymi ogniwami, a prawdziwe baterie mają większe rozmiary, korzystamy z nich np. do zasilania latarek. Wśród ogniw wykorzystywanych w charakterze źródeł prądu wyróżniamy ogniwa regenerowalne oraz ogniwa nieregenerowalne. W przypadku ogniw nieregenerowalnych reakcje zachodzące na elektrodach wywołują trwałe zmiany chemiczne. Po wykorzystaniu takie ogniwo jest bezużyteczne. Inaczej jest w przypadku ogniw regenerowalnych zwanych akumulatorami. Można z nich korzystać wiele razy, ponieważ reakcje elektrodowe można odwrócić i doprowadzić układ do pierwotnego stanu. Aby tego dokonać należy podłączyć ogniwo do innego źródła prądu o odpowiednim napięciu. Zachodzą wówczas takie same procesy jak w czasie pracy ogniwa, lecz ich kierunek jest przeciwny.Tak więc to co na danej elektrodzie było substratem staje się produktem i odwrotnie - to co było produktem staje się substratem. Proces odwracania reakcji zachodzących w ogniwie nazywamy ładowaniem a w ujęciu elektrochemii to nic innego jak elektroliza. A więc rozładowanie akumulatora nie prowadzi do jego zniszczenia, gdyż zawsze można go ponownie naładować kosztem prądu pochodzącego z zewnętrznego źródła energii elektrycznej.
Typowym nieregenerowalnym ogniwem jest ogniwo Leclanche, które można opisać schematem:
(-) Zn | NH4Cl(aq) | MnO2, C (+)
(indeks aq dotyczy całej cząsteczki chlorku amonu i oznacza, że związek ten występuje w postaci wodnego roztworu)
W tym przypadku anodą jest cynk, ulega on więc utlenieniu (dla przypomnienia - na anodzie zawsze zachodzi proces utleniania). Z kolei na katodzie, w postaci pręta węglowego, następuje redukcja dwutlenku manganu. Kubek cynkowy wypełniony jest zagęszczonym mąką ziemniaczaną roztworem salmiaku (chlorku amonu) o stężeniu około 20%. Całość otoczona jest woreczkiem zbudowanym z depolaryzatora, którym jest dwutlenek manganu. W czasie pobierania prądu przez urządzenie elektryczne salmiak (NH4Cl) ulega elektrolizie. Powstałe w ten sposób jony naładowane ujemnie wędrują do anody (blaszka cynkowa), oddają tam ładunek elektryczny, ulegają zobojętnieniu i wchodzą w reakcję z cynkiem tworząc nowy związek chemiczny. Dodatnio naładowane jony wodorowe wędrują w kierunku drugiej elektrody (węglowej). Żeby zapobiec powstawaniu pęcherzyków wodoru tworzących się w wyniku zobojętniania protonów, wokół pręta węglowego umieszcza się depolaryzator w postaci ditlenku manganu, który redukuje się utleniając tym samym wodór. Ogniwo to będzie pracować dopóki będą zachodzić reakcje z cynkiem, później proces zostaje nieodwracalnie zahamowany.
Zachodzące reakcje można zapisać sumarycznie:
4 MnO2 + 4 NH4Cl + 2 Zn → 4 MnO(OH) + ZnCl2 + [Zn(NH3)4]Cl2
Ogniwo Leclanchego generuje napięcie równe w przybliżeniu 1,5 V, które powoli maleje w miarę zużywania ogniwa. Jego konstrukcja jest stale udoskonalana tak, by można było czerpać więcej energii przez dłuższy czas.
Akumulatory są specjalnymi ogniwami elektrolitycznymi, które mogą służyć jako źródło prądu jeśli się je naładuje, tzn. przepuści się przez nie prąd stały przez pewien określony czas. W trakcie ładowania przebiega elektroliza danego elektrolitu, jednocześnie dochodzi do zmian chemicznych w warstwie tzw. mas czynnych na płytach akumulatora. Tak więc można odwrócić reakcje chemiczne bez konieczności zamiany elektrod, a jedynie poprzez przepuszczenie przez odwracalne ogniwo prądu w kierunku przeciwnym. Ładowanie jest niczym innym jak zamienianiem energii elektrycznej dostarczanej przez źródło prądu na energię chemiczną. Zmagazynowana w akumulatorze energia chemiczna będzie mogła ulegać przekształceniu z powrotem na energię elektryczną podczas rozładowywania.
Jednym z parametrów charakteryzujących akumulatory jest pojemność, która jest miarą ilości ładunku elektrycznego jaki można uzyskać w wyniku całkowitego rozładowania akumulatora. Przykładowo pojemność 12-godzinna oznacza, że akumulator może być rozładowywany ciągle przez 12 godzin. Innym parametrem jest sprawność akumulatora definiowana jako iloraz całkowitego ładunku elektrycznego jaki otrzymuje się w wyniku pobierania prądu i ilości energii potrzebnej do jego naładowania. Zależy ona w dużej mierze od rodzaju akumulatora. Przeciętna sprawność to w przybliżeniu 80%.
W trakcie ładowania elektrolit nagrzewa się i oddaje ciepło. Wyższe temperatury elektrolitu wpływają niekorzystnie na płyty akumulatora obniżając ich trwałość. Stąd też ważne jest, aby w czasie ładowania stale kontrolować temperaturę elektrolitu. Jeśli osiągnie ona poziom 40°C to należy przerwać proces ładowania na jakiś czas.
Najczęściej korzysta się z dwóch podstawowych typów akumulatorów - kwasowych i zasadowych, przy czym wśród kwasowych najpopularniejsze są akumulatory ołowiowe, a wśród zasadowych - żelazowo-niklowe.
Na akumulator ołowiowy składają się dwie płyty zanurzone w elektrolicie, czyli 37 procentowym wodnym roztworze kwasu siarkowego (VI). Do prawidłowej pracy akumulatora wymagana jest gęstość elektrolitu granicach (1,27 - 1,29) g/cm3. Budowa i powierzchnia płyt akumulatora determinują jego pojemność i maksymalne natężenie prądu, jakie może być wytwarzane. Płyty zazwyczaj zrobione są z ołowianych krat, które wypełnia tzw. masa czynna o porowatej strukturze. Można je rozróżnić po kolorze - w naładowanym akumulatorze płyta dodatnia ma zabarwienie brunatne pochodzące od dwutlenku ołowiu, a płyta ujemna jest szara od porowatego ołowiu metalicznego.
Cały układ elektrochemiczny umieszczony jest w szklanym albo ebonitowym naczyniu. Zamieszcza się w nim wiele płyt, aby zwiększyć pojemność. Płyty dodatnie przedzielone są płytami ujemnymi, przy czym ujemnych płyt jest więcej. Jest tak dlatego, gdyż szereg elektrod powinien być z obu stron zakończony płytami ujemnymi ze względu na to, że są one odporniejsze na wszelkie uszkodzenia. Elektrody o tym samym znaku łączy się ze sobą za pomocą ołowianych mostków. Płyty o przeciwnych biegunach nie mogą się ze sobą stykać. Aby spełnić ten warunek stosuje się bariery w postaci szklanych rurek albo nie przewodzących prądu przekładek porowatych nasiąkniętych elektrolitem.
Przemiany chemiczne zachodzące podczas pracy akumulatora prowadzą do przekształcania się masy czynnej tak, że stopniowo elektrody coraz bardziej pokrywają się siarczanem (VI) ołowiu (II), czyli PbSO4. Jednocześnie zmniejszeniu ulega gęstość elektrolitu (jony siarczanowe wykorzystywane są na budowę nowego związku chemicznego, a nowe porcje jonów nie są dostarczane przez układ, dodatkowo w wyniku reakcji powstaje woda). Gęstość elektrolitu jest wskaźnikiem stopnia naładowania i wskazuje kiedy należy doładować akumulator. Granicą jest gęstość 1,19 g/cm3 - poniżej tej wartości trzeba naładować akumulator. W trakcie ładowania dochodzi do przemiany PbSO4 odpowiednio w PbO2 na elektrodzie dodatniej i w porowaty ołów na elektrodzie ujemnej. Równocześnie do elektrolitu powracają jony kwasu siarkowego, a więc zwiększa się jego gęstość. Średnio jedno ogniwo w akumulatorze ołowiowym ma napięcie ok. 2 V. Postępujący proces rozładowania powoduje spadek napięcia. Im większe natężenie pobieranego prądu, tym napięcie spada szybciej.
Jeśli napięcie spadnie do poziomu ok. 1,8 V to trzeba przerwać korzystanie z akumulatora i w jak najszybszym czasie poddać go ładowaniu - inaczej może dojść do uszkodzenia akumulatora. Ładowanie akumulatora wymaga większej energii, niż jest to określone przez pojemność, ponieważ pewna część energii zużywana jest na pokonanie oporu wewnętrznego a także na uzupełnienie wewnętrznych strat cieplnych i chemicznych.
Ten typ akumulatorów stosowany jest przede wszystkim samochodach i motocyklach.
W akumulatorach zasadowych jako elektrolit służy zasada potasowa. Obok żelazowo-niklowych stosuje się też akumulatory kadmowo-żelazowo- niklowe.
W tych akumulatorach procesy związane z ładowaniem i rozładowaniem są bardziej złożone niż w przypadku ołowiowych akumulatorów.
W odniesieniu do akumulatorów ołowiowych, akumulatory zasadowe są lżejsze, bardziej odporne na wstrząsy, zwarcia, zmiany napięcia. Poza tym mogą pozostawać przez jakiś czas całkowicie rozładowane bez szkody dla układu galwanicznego. Ich obsługiwanie także jest prostsze. Nie są jednak pozbawione wad. Po pierwsze akumulatory zasadowe mają mniejszą siłę elektromotoryczną, po drugie - charakteryzują się kilkakrotnie większym oporem wewnętrznym co oznacza, że w sumie są mniej wydajne (ich sprawność zazwyczaj nie przekracza 50%). Na ich niekorzyść przemawia też ich wysoka cena.
Dzisiejszy świat byłby dużo uboższy bez ogniw elektrochemicznych. Ich obecność bardzo ułatwia wiele aspektów życia współczesnego człowieka.
KOROZJA I METODY JEJ ZAPOBIEGANIA
Pojęcie korozji jest dość szerokie i obejmuje większość procesów związanych z niszczeniem jakiegoś tworzywa pod wpływem działania środowiska. Najczęściej korozja kojarzy nam się z ubytkami w warstwie metali lub stopów. Rzeczywiście to zjawisko dotyczy przede wszystkim przedmiotów metalicznych, ale należy pamiętać, że korozja może dotykać także wszelkich niemetalicznych tworzyw, takich jak beton, ceramika czy tworzywa sztuczne. Ponieważ otacza nas bardzo dużo przedmiotów wykonanych z tych materiałów, korozja jest poważnym problemem dla gospodarki i corocznie jest przyczyną ogromnych strat.
Każdy widział kiedyś rdzę ale czy wiemy czym ona jest w istocie? Rozpatrując ją pod względem chemicznym stwierdzamy, że nie jest jednolitą, określoną substancją, tylko dość niejednoznaczną kombinacją różnych związków mających w swym składzie pierwiastki takie jak żelazo, tlen i wodór. Głównym składnikiem rdzy są związki żelaza na +3 stopniu utlenienia - tlenki, węglany i wodorotlenki.
Skuteczne przeciwdziałanie korozji wymaga dokładnego poznania mechanizmu i przyczyn jej powstawania. Z codziennej, dość powierzchownej obserwacji otoczenia można wnioskować, że do powstania rdzy np. na żelazie rzadko wystarcza obecność wody czy tlenu.Oprócz tych związków istnieje czynnik znacznie przyspieszający proces rdzewienia żelaza - są to jony wodorowe H+. Informacje te umożliwiają ustalenie prawdopodobnego mechanizmu powstawania rdzy. Na początku zwilżenie wodą powierzchni żelaza powoduje jego częściowe zjonizowanie, które możemy przedstawić w postaci równania:
Fe → Fe2+ + 2e-
Znajdujące się w pobliżu jony wodorowe wyłapują elektrony i tym samym przesuwają równowagę powyższej reakcji w prawo.
2e- + 2H+ → H2
Ze względu na to, że podczas rdzewienia nie dochodzi do wydzielania gazowego wodoru i biorąc pod uwagę, że w beztlenowych warunkach korozji nie spotyka się, można wnioskować że zobojętnione atomy wodoru zamiast tworzyć cząsteczkowy gaz, reagują z tlenem wg równania:
4H + O2 →2 H2O
W ten sposób usuwany jest z układu wodór, a więc znów równowaga przesuwana jest w prawo.
Podany wyżej schemat nie uwzględnia wszystkich przyczyn, jakie wpływają na szybkość zachodzenia korozji. Poza tym odnosi się do sytuacji, w której żelazo jest czystym metalem, co w praktyce zdarza się raczej rzadko - albo żelazo samo w sobie ma domieszki innych pierwiastków albo jego powierzchnia jest zanieczyszczona drobinami osadzającymi się z powietrza lub wody deszczowej. Obecność w pobliżu innych substancji w istotny sposób wpływa na przebieg korozji. Można to zaobserwować w prostych doświadczeniach. 1) Zupełnie czysty pod względem chemicznym cynk wrzucamy do probówki, dodajemy kwasu solnego albo siarkowego. Obserwujemy bardzo powolne rozpuszczanie się metalu i ledwie dostrzegalne pęcherzyki gazu. Całkiem inaczej będzie zachowywał się tzw. cynk techniczny, który nie jest idealnie czysty. W tym przypadku zaobserwujemy energiczną, bardzo szybko przebiegającą reakcję stale zwiększającą swoją szybkość. Podobne różnice można zaobserwować w czasie reakcji z kwasem technicznego i czystego chemicznie żelaza.
2) Analogiczne zwiększenie szybkości reakcji obserwuje się w sytuacji, gdy czysty cynk lub żelazo połączy się z kawałkiem metalu, który jest bardziej szlachetny, może to być np. miedź. Dodanie kwasu do takiego układu powoduje intensywne rozpuszczanie się cynku lub żelaza.
Te dwa zjawiska da się wytłumaczyćpowstawaniem elektrochemicznych ogniw. Mniej szlachetny metal stanowi anodę, a substancja mająca bardziej dodatni potencjał normalny stanowi katodę. Ze względu na to, że w ogniwie elektroda ujemna (anoda) zawsze ulega rozpuszczeniu, nie należy się dziwić, że korozja dotyka metalu mniej szlachetnego. Cynk techniczny zazwyczaj zawiera domieszki innych pierwiastków takich jak miedź, srebro czy węgiel, które kontaktując się z roztworem elektrolitu i cynkiem powodują tworzenie się mikroogniw. W przypadku żelaza często dodatni biegun mikroogniwa tworzą małe skupiska grafitu, siarczków lub tlenków żelaza. W trakcie rozpuszczania metalu powstaje coraz więcej podobnych domieszek, które osiągają coraz większe zagęszczenie, ilość mikroogniw wzrasta - proces korozji ulega przyspieszeniu i intensyfikacji.
Typy i rodzaje korozji:
-Korozja elektromechaniczna; najpopularniejszy typ korozji dotykający metali. Spowodowana jest niejednolitą strukturą i tworzeniem się różnic potencjałów w zewnętrznych warstwach metalu.Podlega identycznym regułom jakie panują w rozważaniach nad reakcjami w ogniwach elektrochemicznych. Stąd też wiele pojęć, określeń i zależności typowych dla ogniw i elektrochemii dobrze opisuje to, co dzieje się w trakcie korozji.
-Korozja chemiczna; jest procesem utleniania metali w środowisku suchych gazów lub cieczy nie wykazujących właściwości elektrolitu (np. ciecze organiczne). Korozję chemiczną charakteryzuje to, że wszystkie procesy (utlenianie, redukcja, powstanie produktu) zachodzą w jednym miejscu na powierzchni metalu przy czym nie występuje swobodny przepływ elektronów poprzez granicę dwóch faz.
-Korozja lokalna; jest to pojęcie stosowane do określenia nierównomiernej korozji metalu w wodnym środowisku. Obszary anodowe można odróżnić od katodowych gołym okiem albo z pomocą mikroskopu. Powierzchnia katody jest dużo większa niż powierzchnia anody. Powstające w wyniku korozji produkty nie stanowią powłoki chroniącej przed dalszym postępem tego procesu. Przekroczenie iloczynu rozpuszczalności powoduje ich wytrącanie się i odkładanie w obszarze między katodą a anodą.
-Korozja ogólna; dotyczy sytuacji, kiedy tworzące się ogniwa są bardzo małe i w związku z tym dochodzi do równomiernej korozji. Cała powierzchnia składa się z miejsc anodowych i katodowych zmieniających co jakiś czas miejsce. Produkty korozji albo przechodzą do rozpuszczalnika albo wydzielają się w równomierny sposób na powierzchni danego metalu. Ten typ korozji elektrochemicznej wywołuje zniszczenia równomierne.
-Korozja atmosferyczna; jak sama nazwa wskazuje związana jest z działaniem na tworzywa substancji zawartych w powietrzu atmosferycznym. To, jak szybko będzie zachodzić zależy od ilości zanieczyszczeń i wilgoci w powietrzu oraz od materiału, tzn. z jakich substancji powstał, jaką ma strukturę, jaką ma powierzchnię itp., itd. Szacuje się, że korozja atmosferyczna zachodzi wtedy, gdy powietrze ma wilgotność względną większą niż 70%, ponieważ przy takiej koncentracji pary wodnej może dojść do jej kondensacji i powstania małych kropelek wody na warstwie jakiegoś materiału (najłatwiej jest w przypadku metalu). Nie bez znaczenia jest także klimat i mikroklimaty występujące w ramach danej strefy klimatycznej. Zanieczyszczenia zwarte w powietrzu, takie jak np. tlenki siarki przyspieszają korozję, ponieważ zwiększają przewodnictwo skroplonej na materiale pary wodnej. To oznacza, że elektrony i jony są przenoszone szybciej i efektywniej co jest równoznaczne ze zwiększeniem intensywności pracy powstałego ogniwa. Z kolei zanieczyszczenia stałe osadzone na powierzchni metalu (zwłaszcza sadza) wpływają na zwiększenie tempa zachodzącej korozji przez przyspieszenie procesu redukcji tlenu na katodzie.
-Korozja galwaniczna; spowodowana jest kontaktem między dwoma różnymi metalami lub stopami o innych potencjałach elektrochemicznych, co jest przyczyną utworzenia się ogniwa. Taka sytuacja następuje tylko wtedy, gdy różnica w potencjałach jest większa niż 50 mV, mniejsze różnice w zasadzie nie mają znaczenia. Im bardziej różnią się od siebie potencjały metali mających styczność ze sobą, tym ogniwo jest efektywniejsze. Za przykład tego typu korozji może posłużyć korozja stali z domieszką węgla mającej styczność ze stalą z domieszka chromu i niklu. Podobny efekt będziemy obserwować w przypadku kontaktu przedmiotu wykonanego z mosiądzu albo miedzi ze stalą tradycyjną albo pokrytą cynkiem, która będzie korodować gdy cynk się rozpuści. Jeśli sytuacja dotyczy metali, które się silnie pasywują, nawet duża różnica potencjałów nie wywoła znaczących efektów korozyjnych. Powodem jest dobra ochrona utworzonych na powierzchni metali warstewek pasywnych (tworzonych przez związki danego metalu - najczęściej tlenki). Praktyczne wykorzystanie wiedzy na temat korozji galwanicznej pozwala na zmniejszenie niepożądanego efektu w sytuacjach, kiedy łączenie metali jest konieczne. W tym celu przestrzega się kilku zasad: należy zadbać o to, żeby metale, które trzeba połączyć miały jak najmniejsza różnicę potencjałów oraz należy dobierać metale w taki sposób, aby metal o większym potencjale (będzie katodą) miał niewielką powierzchnię, natomiast metal o niższym potencjale (będzie anodą) - stosunkowo dużą. W ten sposób zmniejsza się szkodliwość efektu korozyjnego przez rozłożenie go na większej przestrzeni. Z tego względu w pracy z konstrukcjami metalowymi powinno się korzystać ze śrub, nakrętek itp. akcesoriów wykonanych z materiałów bardziej szlachetnych niż te, z których wykonane są te konstrukcje. Korozja galwaniczna zachodząca w roztworach wykazujących niewielkie przewodnictwo (np. miękka woda) jest bardziej niebezpieczna niż w przypadku roztworów o większym przewodnictwie, gdyż efekt korozyjny skupia się w pobliżu połączenia metali. Wyżej wymienione przykłady można zaliczyć do makroogniw, ale korozja galwaniczna nie ogranicza się tylko do nich - obejmuje także mikroogniwa. Takie ogniwa w skali mikro spotykamy w ramach pojedynczego fragmentu metalu. Jako przykład może posłużyć korozja selektywna w obrębie stopów, która polega na powstawaniu ubytku substancji mniej szlachetnej (przykładowo w mosiądzu ubywa cynku). Podobnie jest w przypadkuselektywnej korozji szarego żeliwa zamoczonego w wodzie lub zagrzebanego w glebie - w efekcie żelazo przechodzi w rdzę, która zawiera pozostały grafit.
-Korozja naprężeniowa; spotykamy się z nią wtedy, gdy do czynników natury elektrochemicznej dochodzą zmiany związane z powstaniem naprężeń mechanicznych. Ten typ korozji może uwidaczniać się w postaci pęknięć międzykrystalicznych lub śródkrystalicznych.
-Korozja zmęczeniowa; zachodzi w przypadku narażenia metalu na powtarzające się naprężenia w agresywnym środowisku. Ujawnia się pod postacią pęknięć w pewnych miejscach danej konstrukcji. Często ten problem dotyka kotłów parowych i urządzeń mających styczność ze środowiskiem morskiej wody.
-Korozja cierna; ulegają jej powierzchnie styczne dwóch szczelnie przylegających do siebie metali, które drgają albo wykonują przesunięcia oscylacyjne.
-Korozja kawitacyjna; spowodowana jest tworzeniem się w cieczach tzw. luk próżniowych na skutek wibracji lub szybkiego ruchu. Problem dotyczy przede wszystkim śrub okrętowych, wirników turbin hydraulicznych i tym podobnych.
-Korozja kontaktowa; zachodzi na łączeniu dwóch odmiennych metali mających różne potencjały.
-Korozja selektywna; spowodowana jest miejscowym zubożeniem stopu w pewien metal, który jest wiązany przez węgiel. Powstałe węgliki wydzielają się na granicy między ziarnami metalu.
-Korozja wżerowa (ang. pitting); występuje głównie w środowisku jonów chlorkowych, powoduje głębokie ubytki wżerne w materiale (najczęściej chodzi tu o metal).
-Korozja wysokotemperaturowa nazywana też gazową; stanowi chemiczny proces utleniania różnych metali w spalinach albo innych warunkach, w których obecna jest siarka, siarkowodór albo któryś z chlorowców. Uwidacznia się jako zniszczenie struktury metalu i zmiana jego mechanicznej wytrzymałości.
-Korozja katastrofalna; jest to skrajny przypadek wysokotemperaturowej korozji - niszczenie metalu jest bardzo szybkie.
-Korozja kwasowa; dotyczy tworzyw nieorganicznych, do których wnika substancja o charakterze kwaśnym i oddziałuje chemicznie z materiałem.
-Korozja siarczanowa; jest to specyficzny rodzaj korozji dotykający cement portlandzki. Jeśli związki tworzące ten cement wystawione są na działanie siarczanów (mogą one być zawarte np. w pyłach czy w wodzie deszczowej) to zachodzą reakcje prowadzące do powstania soli Candlota. Sole te mają dużą objętość właściwą i dlatego rozsadzają beton.
-Pęcznienie; to zjawisko dotyczy głównie tworzyw zbudowanych ze związków organicznych lub wszelkiego rodzaju plastików. Ich struktura jest stosunkowo luźna, co sprzyja wnikaniu w głąb tworzywa obcych drobin wywołujących pęcznienie i tym samym pogorszenie właściwości mechanicznych materiału.
-Solwatacja;. zachodzi w plastikach. W wyniku reakcji drobin tworzywa z wnikającymi do środka cząsteczkami ze środowiska zewnętrznego może dochodzić do destrukcji objawiającej się tworzeniem kompleksów (solwatów) albo utlenieniem tworzywa.
Jak chronić metale przed korozją?
Z uwagi na potężne starty, które ponosi człowiek z powodu występowania korozji, należało opracować jakieś metody zapobiegania lub chociaż spowalniania tego szkodliwego procesu. W tej chwili znanych jest już wiele sposobów na ochronę antykorozyjną różnorodnych materiałów. Wśród najważniejszych są:
- elektrochemiczna ochrona protektorowa i katodowa;
- ochronne powłoki (metaliczne oraz niemetaliczne);
- ulepszanie powierzchni metali metoda dyfuzyjną;
- inhibitory korozji.
Katodowa ochrona polega na przyłączeniu do fragmentów konstrukcji metalowych narażonych na działanie
korozji bieguna ujemnego źródła energii elektrycznej (prąd stały) o niedużym napięciu rzędu 1 do 2 V. Bardzo często korzysta się z elektrochemicznej ochrony protektorowej, która polega na połączeniu chronionego metalu (np. żelaza) z dużym blokiem metalu o mniejszym potencjale elektrochemicznym - dla żelaza dobry będzie cynk lub magnez. Jeśli obydwa metale znajdą się w jednakowym elektrolicie, to powstanie ogniwo. Reakcje w tym ogniwie będą prowadzić do rozpuszczania bloku magnezu lub cynku a nie żelaza.Metodę ta stosuje się do ochrony podziemnych rurociągów oraz kadłubów statków. Mimo, że wykorzystane całkowicie bloki anodowe muszą być co jakiś czas wymieniane, to jest to dużo łatwiejsze i bardziej ekonomiczne niż ciągłe zasilanie prądem konstrukcji wymagających ochrony (a tak jest w przypadku ochrony katodowej).
Powłoki stworzone z metalu, który jest mniej szlachetny od żelaza obok izolacji od wilgoci i tlenu zapewniają jednocześnie protektorową ochronę. Oznacza to, że nawet jeśli dojdzie do poważnej destrukcji powłoki cynkowej, która została naniesiona stal wymagającą ochrony, to chroniony metal będzie wykazywał ograniczoną podatność na korozję (będzie spełniał rolę katody a nie anody).
Jeżeli powłoka metalowa jest zbudowana z metalu bardziej szlachetnego niż ten, który pokrywa to ochrona jest znacznie mniej skuteczna. Mało tego, w pewnych przypadkach okazuje się, że taka powłoka nie dość że nie chroni to jeszcze wzmaga korozję! Rozważmy przykład płytki żelaznej, którą trzeba zabezpieczyć przed korozją. Metalami bardziej szlachetnymi niż żelazo (mającymi wyższy potencjał standardowy, czyli stojącymi za żelazem w szeregu napięciowym) są np. miedź, cynk czy też nikiel. Jeśli naniesiemy galwanicznie któryś z tych metali na żelazo to taka powłoka będzie spełniać swoje zadanie tylko wtedy, gdy będzie całkowicie szczelna. W momencie uszkodzenia, jeśli w pobliżu będzie wilgoć (wiadomo, że powietrze niemal zawsze zawiera parę wodną) i zanieczyszczenia to natychmiast żelazo zacznie korodować z szybkością większą niż gdyby nie było niczym pokryte. Dzieje się tak dlatego, że zawsze metal o wyższym potencjale będzie tworzył katodę. Tak więc w wyniku uszkodzenia powłoki dochodzi do powstania mikroogniw, w których żelazo pełni rolę anody. Oznacza to, że żelazo będzie się utleniać do kationów Fe2+. Powstające elektrony będą wykorzystywane do redukcji wodoru jeśli elektrolit był kwaśny (jonów H+ dostarczają chociażby kwaśne deszcze) lub do redukcji tlenu w przypadku elektrolitu obojętnego lub zasadowego. Zużywanie elektronów powoduje, że proces będzie zachodził szybciej i dłużej. Żelazo w postaci jonów będzie łatwo reagować z innymi substancjami a w warstwie metalicznej powstaną powiększające się ubytki.
Powłoki niemetaliczne mają za zadanie przede wszystkim izolować powierzchnię metalu tak, żeby nie miała ona styczności ani z tlenem, ani z wilgocią. Najczęściej do tego celu wykorzystuje się kolorowe lakiery i farby, które poza funkcją ochronną mogą też spełniać funkcje zdobnicze. Znane są też inne, skuteczniejsze metody uzyskiwania powłok szczelnych, trwałych i znakomicie przylegających do powierzchni chronionego materiału. Najważniejszą jest pasywacja, czyli utlenianie cieniutkiej warstwy metalu na jego powierzchni. Są takie metale, które same, bez ingerencji człowieka, utleniają się na powietrzu i tworzą na swojej powierzchni cienką lecz zwartą warstwę tlenku. Ponieważ proces ten zachodzi szybko na całej powierzchni metalu, wewnętrzne warstwy są chronione przed postępującą korozją przez tlenki, których ludzkie oko nie dostrzega. Takim metalem, który sam siebie ochrania jest m.in. glin. Stosując odpowiednie zabiegi elektrochemiczne (anodowe utlenianie metalu) uzyskuje się warstwę tlenków o większej grubości i lepszych właściwościach ochronnych.
Do najnowszych metod ochrony metali należy wytwarzanie specjalnych powierzchni stopowych, którymi powleka się powierzchnie metali.W znacznym stopniu przypomina to galwaniczny sposób powlekania metali, z tym że w tym procesie nie używa się wody - rozpuszczalnikami są fluorki metali lekkich do których dodaje się mniej więcej 1% fluorku tego metalu, który tworzy zewnętrzną warstwę stopu. Najważniejszym elementem procesu jest dyfundowanie cząsteczek metalu będącego anodą w głąb cząsteczek znajdujących się na powierzchni metalu drugiego, stanowiącego katodę. Sól fluorkową podgrzewa się do temperatur rzędu 800-1600 kalwinów, w tych warunkach wytwarza się stop powierzchniowy mający grubość średnio od około 25 nawet do kilkuset mm (mikrometrów).Do tej pory udało się uzyskać więcej niż 500 rozmaitych struktur tego typu. Zaliczamy do nich między innymi odporne na procesy korozyjne, dyfuzyjne berylowe powłoki pokrywające przedmioty miedziane, tytanowe, niklowe, kobaltowe, żelazowe, i inne. Dodatkowo powłoki tytanowe na stalach zaliczanych do miękkich, a także na stopach miedziowych i niklowych poprawiają odporność tych materiałów na żrące działanie różnych kwasów. Bardzo korzystne jest też aluminiowanie w środowisku stopionych fluorków. Zaletą stosowania tej metody ochrony antykorozyjnej jest fakt, że można ją stosować nie tylko do metali. Dowodem na to jest uzyskanie twardych i błyszczących powierzchni krzemowych na platynie.
Kolejnym sposobem na zahamowanie zjawiska korozji jest stosowanie inhibitorów, które są substancjami silnie wiążącymi się z powierzchnią metalu. W ten sposób blokowany jest kontakt z jonami wodorowymi. Przykładowo jeśli doda się siarczek difenyloetanu w ilości około 0,05%, to obserwuje się spadek ubytku żelaza w roztworze HCl nawet o 75%. Dzięki inhibitorom można było zrezygnować z natłuszczania cennych i precyzyjnych urządzeń metalowych w celu ich konserwacji czy transportu. Zamiast tego stosuje się szczelne opakowania z folii PE (polietylenowej) zawierające wewnątrz niewielką ilość inhibitorów, które charakteryzują się dużą lotnością. Inhibitory można też dodawać do lakierów i farb, co w znaczny sposób poprawia ich antykorozyjne właściwości .
BIBLIOGRAFIA
1. K.M. Pazdro "Elektrochemia"
2. Multimedialna Encyklopedia PWN
3. "Chemia. Encyklopedia popularna"
4. "Chemia. Encyklopedia ilustrowana"
5. K.M. Pazdro "Podstawy chemii dla kandydatów na wyższe uczelnie"
6. Źródła internetowe
